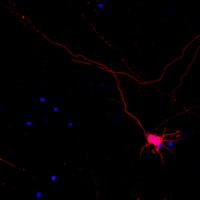
TUBB5 and its disease-associated mutations influence the terminal differentiation and dendritic spine densities of cerebral cortical neurons.
Ngo, L; Haas, M; Qu, Z; Li, SS; Zenker, J; Teng, KS; Gunnersen, JM; Breuss, M; Habgood, M; Keays, DA; Heng, JI
Human molecular genetics
23
5147-58
2014
Show Abstract
The microtubule cytoskeleton is critical for the generation and maturation of neurons in the developing mammalian nervous system. We have previously shown that mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities in humans. While it is known that TUBB5 is necessary for the proper generation and migration of neurons, little is understood of the role it plays in neuronal differentiation and connectivity. Here, we report that perturbations to TUBB5 disrupt the morphology of cortical neurons, their neuronal complexity, axonal outgrowth, as well as the density and shape of dendritic spines in the postnatal murine cortex. The features we describe are consistent with defects in synaptic signaling. Cellular-based assays have revealed that TUBB5 substitutions have the capacity to alter the dynamic properties and polymerization rates of the microtubule cytoskeleton. Together, our studies show that TUBB5 is essential for neuronal differentiation and dendritic spine formation in vivo, providing insight into the underlying cellular pathology associated with TUBB5 disease states. | 24833723
 |
Unc5C and DCC act downstream of Ctip2 and Satb2 and contribute to corpus callosum formation.
Srivatsa, S; Parthasarathy, S; Britanova, O; Bormuth, I; Donahoo, AL; Ackerman, SL; Richards, LJ; Tarabykin, V
Nature communications
5
3708
2014
Show Abstract
The pyramidal neurons of the mammalian neocortex form two major types of long-range connections-corticocortical and cortico-subcortical. The transcription factors Satb2 and Ctip2 are critical regulators of neuronal cell fate that control interhemispheric versus corticofugal connections respectively. Here, we investigate the axon guidance molecules downstream of Satb2 and Ctip2 that establish these connections. We show that the expression of two Netrin1 receptors- DCC and Unc5C is under direct negative regulation by Satb2 and Ctip2, respectively. Further, we show that the Netrin1-Unc5C/DCC interaction is involved in controlling the interhemispherical projection in a subset of early born, deep layer callosal neurons. | 24739528
|
Astrocytes refine cortical connectivity at dendritic spines.
Risher, WC; Patel, S; Kim, IH; Uezu, A; Bhagat, S; Wilton, DK; Pilaz, LJ; Singh Alvarado, J; Calhan, OY; Silver, DL; Stevens, B; Calakos, N; Soderling, SH; Eroglu, C
eLife
3
2014
Show Abstract
During cortical synaptic development, thalamic axons must establish synaptic connections despite the presence of the more abundant intracortical projections. How thalamocortical synapses are formed and maintained in this competitive environment is unknown. Here, we show that astrocyte-secreted protein hevin is required for normal thalamocortical synaptic connectivity in the mouse cortex. Absence of hevin results in a profound, long-lasting reduction in thalamocortical synapses accompanied by a transient increase in intracortical excitatory connections. Three-dimensional reconstructions of cortical neurons from serial section electron microscopy (ssEM) revealed that, during early postnatal development, dendritic spines often receive multiple excitatory inputs. Immuno-EM and confocal analyses revealed that majority of the spines with multiple excitatory contacts (SMECs) receive simultaneous thalamic and cortical inputs. Proportion of SMECs diminishes as the brain develops, but SMECs remain abundant in Hevin-null mice. These findings reveal that, through secretion of hevin, astrocytes control an important developmental synaptic refinement process at dendritic spines. | 25517933
|
Neuron glia-related cell adhesion molecule (NrCAM) promotes topographic retinocollicular mapping.
Dai, J; Buhusi, M; Demyanenko, GP; Brennaman, LH; Hruska, M; Dalva, MB; Maness, PF
PloS one
8
e73000
2013
Show Abstract
NrCAM (Neuron-glial related cell adhesion molecule), a member of the L1 family of cell adhesion molecules, reversibly binds ankyrin and regulates axon growth, but it has not been studied for a role in retinotopic mapping. During development of retino-collicular topography, NrCAM was expressed uniformly in retinal ganglion cells (RGCs) along both mediolateral and anteroposterior retinal axes, and was localized on RGC axons within the optic tract and superior colliculus (SC). Anterograde tracing of RGC axons in NrCAM null mutant mice at P10, when the map resembles its mature form, revealed laterally displaced ectopic termination zones (eTZs) of axons from the temporal retina, indicating defective mediolateral topography, which is governed by ephrinB/EphBs. Axon tracing at P2 revealed that interstitial branch orientation of ventral-temporal RGC axons in NrCAM null mice was compromised in the medial direction, likely accounting for displacement of eTZs. A similar retinocollicular targeting defect in EphB mutant mice suggested that NrCAM and EphB interact to regulate mediolateral retino-collicular targeting. We found that EphB2 tyrosine kinase but not an EphB2 kinase dead mutant, phosphorylated NrCAM at a conserved tyrosine residue in the FIGQY ankyrin binding motif, perturbing ankyrin recruitment in NrCAM transfected HEK293 cells. Furthermore, the phosphorylation of NrCAM at FIGQY in SC was decreased in EphB1/3 and EphB1/2/3 null mice compared to WT, while phospho-FIGQY of NrCAM in SC was increased in EphB2 constitutively active (F620D/F620D) mice. These results demonstrate that NrCAM contributes to mediolateral retinocollicular axon targeting by regulating RGC branch orientation through a likely mechanism in which ephrinB/EphB phosphorylates NrCAM to modulate linkage to the actin cytoskeleton. | 24023801
|
ADAM17 is critical for multipolar exit and radial migration of neuronal intermediate progenitor cells in mice cerebral cortex.
Li, Q; Zhang, Z; Li, Z; Zhou, M; Liu, B; Pan, L; Ma, Z; Zheng, Y
PloS one
8
e65703
2013
Show Abstract
The radial migration of neuronal progenitor cells is critical for the development of cerebral cortex layers. They go through a critical step transforming from multipolar to bipolar before outward migration. A Disintegrin and Metalloprotease 17 (ADAM17) is a transmembrane protease which can process many substrates involved in cell-cell interaction, including Notch, ligands of EGFR, and some cell adhesion molecules. In this study, we used in utero electroporation to knock down or overexpress ADAM17 at embryonic day 14.5 (E14.5) in neuronal progenitor cells to examine the role of ADAM17 in cortical embryonic neurogenesis. Our results showed that the radial migration of ADAM17-knocked down cells were normal till E16.5 and reached the intermediate zone (IZ). Then most transfected cells stopped migration and stayed at the IZ to inner cortical plate (CP) layer at E18.5, and there was higher percentage of multipolar cells at IZ layer in the ADAM17-knocked down group compared to the cells in control group. Marker staining revealed that those ADAM17-knocked down cells differentiated normally from neural stem cells (NSCs) to neuronal intermediate progenitor cells (nIPCs) but did not differentiate into mature neurons. The migration and multipolar exit defects caused by ADAM17 knockdown could be partially rescued by over-expressing an shRNA resistant ADAM17, while overexpressing ADAM17 alone did not affect the radial migration. Taken together, our results showed for the first time that, ADAM17 is critical in regulating the multipolar-stage exit and radial migration of the nIPCs during telencephalon cortex development in mice. | 23755270
|
Evidence that descending cortical axons are essential for thalamocortical axons to cross the pallial-subpallial boundary in the embryonic forebrain.
Chen, Y; Magnani, D; Theil, T; Pratt, T; Price, DJ
PloS one
7
e33105
2012
Show Abstract
Developing thalamocortical axons traverse the subpallium to reach the cortex located in the pallium. We tested the hypothesis that descending corticofugal axons are important for guiding thalamocortical axons across the pallial-subpallial boundary, using conditional mutagenesis to assess the effects of blocking corticofugal axonal development without disrupting thalamus, subpallium or the pallial-subpallial boundary. We found that thalamic axons still traversed the subpallium in topographic order but did not cross the pallial-subpallial boundary. Co-culture experiments indicated that the inability of thalamic axons to cross the boundary was not explained by mutant cortex developing a long-range chemorepulsive action on thalamic axons. On the contrary, cortex from conditional mutants retained its thalamic axonal growth-promoting activity and continued to express Nrg-1, which is responsible for this stimulatory effect. When mutant cortex was replaced with control cortex, corticofugal efferents were restored and thalamic axons from conditional mutants associated with them and crossed the pallial-subpallial boundary. Our study provides the most compelling evidence to date that cortical efferents are required to guide thalamocortical axons across the pallial-subpallial boundary, which is otherwise hostile to thalamic axons. These results support the hypothesis that thalamic axons grow from subpallium to cortex guided by cortical efferents, with stimulation from diffusible cortical growth-promoting factors. | 22412988
|
EphB regulates L1 phosphorylation during retinocollicular mapping.
Jinxia Dai,Jasbir S Dalal,Sonal Thakar,Mark Henkemeyer,Vance P Lemmon,Jill S Harunaga,Monika C Schlatter,Mona Buhusi,Patricia F Maness
Molecular and cellular neurosciences
50
2012
Show Abstract
Interaction of the cell adhesion molecule L1 with the cytoskeletal adaptor ankyrin is essential for topographic mapping of retinal ganglion cell (RGC) axons to synaptic targets in the superior colliculus (SC). Mice mutated in the L1 ankyrin-binding motif (FIGQY(1229)H) display abnormal mapping of RGC axons along the mediolateral axis of the SC, resembling mouse mutant phenotypes in EphB receptor tyrosine kinases. To investigate whether L1 functionally interacts with EphBs, we investigated the role of EphB kinases in phosphorylating L1 using a phospho-specific antibody to the tyrosine phosphorylated FIGQY(1229) motif. EphB2, but not an EphB2 kinase dead mutant, induced tyrosine phosphorylation of L1 at FIGQY(1229) and perturbed ankyrin recruitment to the membrane in L1-transfected HEK293 cells. Src family kinases mediated L1 phosphorylation at FIGQY(1229) by EphB2. Other EphB receptors that regulate medial-lateral retinocollicular mapping, EphB1 and EphB3, also mediated phosphorylation of L1 at FIGQY(1229). Tyrosine(1176) in the cytoplasmic domain of L1, which regulates AP2/clathrin-mediated endocytosis and axonal trafficking, was not phosphorylated by EphB2. Accordingly mutation of Tyr(1176) to Ala in L1-Y(1176)A knock-in mice resulted in normal retinocollicular mapping of ventral RGC axons. Immunostaining of the mouse SC during retinotopic mapping showed that L1 colocalized with phospho-FIGQY in RGC axons in retinorecipient layers. Immunoblotting of SC lysates confirmed that L1 was phosphorylated at FIGQY(1229) in wild type but not L1-FIGQY(1229)H (L1Y(1229)H) mutant SC, and that L1 phosphorylation was decreased in the EphB2/B3 mutant SC. Inhibition of ankyrin binding in L1Y(1229)H mutant RGCs resulted in increased neurite outgrowth compared to WT RGCs in retinal explant cultures, suggesting that L1-ankyrin binding serves to constrain RGC axon growth. These findings are consistent with a model in which EphB kinases phosphorylate L1 at FIGQY(1229) in retinal axons to modulate L1-ankyrin binding important for mediolateral retinocollicular topography. | 22579729
|
Neuronal cadherin (NCAD) increases sensory neurite formation and outgrowth on astrocytes.
Ferguson, TA; Scherer, SS
Neuroscience letters
522
108-12
2012
Show Abstract
We examined the neurite outgrowth of sensory neurons on astrocytes following the genetic deletion of N-cadherin (NCAD). Deletion abolished immunostaining for NCAD and the other classical cadherins, indicating that NCAD is likely the only classical cadherin expressed by astrocytes. Only 38% of neurons grown on NCAD-deficient astrocytes for 24 h produced neurites, as compared to 74% of neurons grown on NCAD-expressing astrocytes. Of the neurons that produced neurites, those grown on NCAD-deficient astrocytes had a mean total length of 378 μm, as compared to 1093 μm for neurons grown on NCAD-expressing astrocytes. Thus, the loss of NCAD greatly impairs the formation and extension neurites on astrocytes. | 22698587
|
Heparan sulfate sugar modifications mediate the functions of slits and other factors needed for mouse forebrain commissure development.
Conway, CD; Howe, KM; Nettleton, NK; Price, DJ; Mason, JO; Pratt, T
The Journal of neuroscience : the official journal of the Society for Neuroscience
31
1955-70
2011
Show Abstract
Heparan sulfate proteoglycans are cell surface and secretory proteins that modulate intercellular signaling pathways including Slit/Robo and FGF/FGFR. The heparan sulfate sugar moieties on HSPGs are subject to extensive postsynthetic modification, generating enormous molecular complexity that has been postulated to provide increased diversity in the ability of individual cells to respond to specific signaling molecules. This diversity could help explain how a relatively small number of axon guidance molecules are able to instruct the extremely complex connectivity of the mammalian brain. Consistent with this hypothesis, we previously showed that mutant mice lacking the heparan sulfotransferases (Hsts) Hs2st or Hs6st1 display major axon guidance defects at the developing optic chiasm. Here we further explore the role of these Hsts at the optic chiasm and investigate their function in corpus callosum development. Each Hst is expressed in a distinct pattern and each mutant displays a specific spectrum of axon guidance defects. Particular Hs2st(-/-) and Hs6st1(-/-) phenotypes closely match those of Slit1(-/-) and Slit2(-/-) embryos respectively, suggesting possible functional relationships. To test functional interactions between Hs2st or Hs6st1 and Slits we examined optic chiasm and corpus callosum phenotypes in a panel of genotypes where Hs2st or Hs6st1 and Slit1 or Slit2 function were simultaneously reduced or absent. We find examples of Hs2st and Hs6st1 having epistatic, synergistic, and antagonistic genetic relationships with Slit1 and/or Slit2 depending on the context. At the corpus callosum we find that Hs6st1 has Slit-independent functions and our data indicate additional roles in FGF signaling. | 21307234
|
Fezf1 and Fezf2 are required for olfactory development and sensory neuron identity.
Eckler, MJ; McKenna, WL; Taghvaei, S; McConnell, SK; Chen, B
The Journal of comparative neurology
519
1829-46
2011
Show Abstract
The murine olfactory system consists of main and accessory systems that perform distinct and overlapping functions. The main olfactory epithelium (MOE) is primarily involved in the detection of volatile odorants, while neurons in the vomeronasal organ (VNO), part of the accessory olfactory system, are important for pheromone detection. During development, the MOE and VNO both originate from the olfactory pit; however, the mechanisms regulating development of these anatomically distinct organs from a common olfactory primordium are unknown. Here we report that two closely related zinc-finger transcription factors, FEZF1 and FEZF2, regulate the identity of MOE sensory neurons and are essential for the survival of VNO neurons respectively. Fezf1 is predominantly expressed in the MOE while Fezf2 expression is restricted to the VNO. In Fezf1-deficient mice, olfactory neurons fail to mature and also express markers of functional VNO neurons. In Fezf2-deficient mice, VNO neurons degenerate prior to birth. These results identify Fezf1 and Fezf2 as important regulators of olfactory system development and sensory neuron identity. | 21452247
|