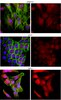
Axonal amyloid precursor protein and its fragments undergo somatodendritic endocytosis and processing.
Niederst, ED; Reyna, SM; Goldstein, LS
Molecular biology of the cell
26
205-17
2015
Zobrazit abstrakt
Deposition of potentially neurotoxic Aβ fragments derived from amyloid precursor protein (APP) at synapses may be a key contributor to Alzheimer's disease. However, the location(s) of proteolytic processing and subsequent secretion of APP fragments from highly compartmentalized, euploid neurons that express APP and processing enzymes at normal levels is not well understood. To probe the behavior of endogenous APP, particularly in human neurons, we developed a system using neurons differentiated from human embryonic stem cells, cultured in microfluidic devices, to enable direct biochemical measurements from axons. Using human or mouse neurons in these devices, we measured levels of Aβ, sAPPα, and sAPPβ secreted solely from axons. We found that a majority of the fragments secreted from axons were processed in the soma, and many were dependent on somatic endocytosis for axonal secretion. We also observed that APP and the β-site APP cleaving enzyme were, for the most part, not dependent on endocytosis for axonal entry. These data establish that axonal entry and secretion of APP and its proteolytic processing products traverse different pathways in the somatodendritic compartment before axonal entry. | | | 25392299
 |
Amyloid-beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies.
Chen, WT; Hong, CJ; Lin, YT; Chang, WH; Huang, HT; Liao, JY; Chang, YJ; Hsieh, YF; Cheng, CY; Liu, HC; Chen, YR; Cheng, IH
PloS one
7
e35807
2011
Zobrazit abstrakt
Amyloid precursor protein (APP) mutations associated with familial Alzheimer's disease (AD) usually lead to increases in amyloid β-protein (Aβ) levels or aggregation. Here, we identified a novel APP mutation, located within the Aβ sequence (Aβ(D7H)), in a Taiwanese family with early onset AD and explored the pathogenicity of this mutation. Cellular and biochemical analysis reveal that this mutation increased Aβ production, Aβ42/40 ratio and prolonged Aβ42 oligomer state with higher neurotoxicity. Because the D7H mutant Aβ has an additional metal ion-coordinating residue, histidine, we speculate that this mutation may promote susceptibility of Aβ to ion. When co-incubated with Zn(2+) or Cu(2+), Aβ(D7H) aggregated into low molecular weight oligomers. Together, the D7H mutation could contribute to AD pathology through a "double punch" effect on elevating both Aβ production and oligomerization. Although the pathogenic nature of this mutation needs further confirmation, our findings suggest that the Aβ N-terminal region potentially modulates APP processing and Aβ aggregation, and further provides a genetic indication of the importance of Zn(2+) and Cu(2+) in the etiology of AD. | | | 22558227
|
Clioquinol reduces zinc accumulation in neuritic plaques and inhibits the amyloidogenic pathway in AβPP/PS1 transgenic mouse brain.
Tao Wang,Chun-Yan Wang,Zhong-Yan Shan,Wei-Ping Teng,Zhan-You Wang
Journal of Alzheimer's disease : JAD
29
2011
Zobrazit abstrakt
Metal dyshomeostasis in the brain helps promote amyloid-β (Aβ) deposition in Alzheimer's disease (AD). Therefore, targeting the interactions between metal and Aβ is a potential therapeutic approach for AD. The metal chelator, clioquinol (CQ), is thought to reduce Aβ deposits in the AD transgenic mouse brain, and attenuate the clinical symptoms of AD patients. However, whether oral administration of CQ reduces zinc accumulation in Aβ plaques and inhibits the amyloidogenic pathway have not been properly established in AD transgenic mice. By means of autometallographic analysis, we show for the first time that both the number and size of the zinc-containing plaques were significantly reduced in the brain of amyloid-β protein precursor (AβPP)/presenilin 1 (PS1) double transgenic mice treated with CQ (30 mg/kg/day) orally for 2 months. This was accompanied by a reduction in Aβ burden in the CQ-treated mouse brain. Furthermore, CQ treatment markedly reduced the expression levels of AβPP protein, the β-site of AβPP cleaving enzyme 1 (BACE1), PS1, and the secreted β-secretase-derived fragments of AβPP (sAβPPβ). The present data indicate that CQ is able to reduce zinc accumulation in the neuritic plaques and inhibit amyloidogenic AβPP processing in the AβPP/PS1 mouse brain. | | | 22269164
|
AβPP intracellular C-terminal domain function is related to its degradation processes.
Erica Buoso,Fabrizio Biundo,Cristina Lanni,Gennaro Schettini,Stefano Govoni,Marco Racchi
Journal of Alzheimer's disease : JAD
30
2011
Zobrazit abstrakt
The amyloid-β protein precursor (AβPP) can be processed by either the amyloidogenic or the non-amyloidogenic pathway; both pathways lead to release of the AβPP intracellular C-terminal domain (AICD). AICD involvement in signal transduction within Fe65/Tip60 complex is one of the most discussed mechanisms, and different models have been hypothesized to explain the role of AICD within this complex. The analysis of these models in relation to the degradation processes highlights the discrepancy among AICD localization, function, and degradation, leading to the hypothesis that a signaling mechanism may exist which allows AβPP proteolysis to generate either a transcriptionally active fragment or an inactive one with different involvement of proteasome and IDE (insulin-degrading enzyme). Our work aimed to analyze the functional role of AICD within the Fe65/Tip60 complex considering the AICD degradation processes. Our data suggest a correlation between the role of AICD in gene regulation and its removal operated by proteasome activity. Moreover, treatments with IDE inhibitor underlined the presence of an alternative mechanism involved in AICD removal when the latter is not exerting nuclear activity, thus providing clearer support for the existence of at least two mechanisms as previously suggested. | | | 22451313
|
Alzheimer's disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging.
Maarouf, CL; Daugs, ID; Kokjohn, TA; Walker, DG; Hunter, JM; Kruchowsky, JC; Woltjer, R; Kaye, J; Castaño, EM; Sabbagh, MN; Beach, TG; Roher, AE
PloS one
6
e27291
2010
Zobrazit abstrakt
The amyloid cascade hypothesis provides an economical mechanistic explanation for Alzheimer's disease (AD) dementia and correlated neuropathology. However, some nonagenarian individuals (high pathology controls, HPC) remain cognitively intact while enduring high amyloid plaque loads for decades. If amyloid accumulation is the prime instigator of neurotoxicity and dementia, specific protective mechanisms must enable these HPC to evade cognitive decline. We evaluated the neuropathological and biochemical differences existing between non-demented (ND)-HPC and an age-matched cohort with AD dementia. The ND-HPC selected for our study were clinically assessed as ND and possessed high amyloid plaque burdens. ELISA and Western blot analyses were used to quantify a group of proteins related to APP/Aβ/tau metabolism and other neurotrophic and inflammation-related molecules that have been found to be altered in neurodegenerative disorders and are pivotal to brain homeostasis and mental health. The molecules assumed to be critical in AD dementia, such as soluble or insoluble Aβ40, Aβ42 and tau were quantified by ELISA. Interestingly, only Aβ42 demonstrated a significant increase in ND-HPC when compared to the AD group. The vascular amyloid load which was not used in the selection of cases, was on the average almost 2-fold greater in AD than the ND-HPC, suggesting that a higher degree of microvascular dysfunction and perfusion compromise was present in the demented cohort. Neurofibrillary tangles were less frequent in the frontal cortices of ND-HPC. Biochemical findings included elevated vascular endothelial growth factor, apolipoprotein E and the neuroprotective factor S100B in ND-HPC, while anti-angiogenic pigment epithelium derived factor levels were lower. The lack of clear Aβ-related pathological/biochemical demarcation between AD and ND-HPC suggests that in addition to amyloid plaques other factors, such as neurofibrillary tangle density and vascular integrity, must play important roles in cognitive failure. | | | 22087282
|
Huperzine A activates Wnt/β-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model.
Wang, CY; Zheng, W; Wang, T; Xie, JW; Wang, SL; Zhao, BL; Teng, WP; Wang, ZY
Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology
36
1073-89
2010
Zobrazit abstrakt
Huperzine A (HupA) is a reversible and selective inhibitor of acetylcholinesterase (AChE), and it has multiple targets when used for Alzheimer's disease (AD) therapy. In this study, we searched for new mechanisms by which HupA could activate Wnt signaling and reduce amyloidosis in AD brain. A nasal gel containing HupA was prepared. No obvious toxicity of intranasal administration of HupA was found in mice. HupA was administered intranasally to β-amyloid (Aβ) precursor protein and presenilin-1 double-transgenic mice for 4 months. We observed an increase in ADAM10 and a decrease in BACE1 and APP695 protein levels and, subsequently, a reduction in Aβ levels and Aβ burden were present in HupA-treated mouse brain, suggesting that HupA enhances the nonamyloidogenic APP cleavage pathway. Importantly, our results further showed that HupA inhibited GSK3α/β activity, and enhanced the β-catenin level in the transgenic mouse brain and in SH-SY5Y cells overexpressing Swedish mutation APP, suggesting that the neuroprotective effect of HupA is not related simply to its AChE inhibition and antioxidation, but also involves other mechanisms, including targeting of the Wnt/β-catenin signaling pathway in AD brain. | | | 21289607
|
Chronic treatment with a novel ?-secretase modulator, JNJ-40418677, inhibits amyloid plaque formation in a mouse model of Alzheimer's disease.
B Van Broeck,J-M Chen,G Tréton,M Desmidt,C Hopf,N Ramsden,E Karran,M Mercken,A Rowley
British journal of pharmacology
163
2010
Zobrazit abstrakt
?-Secretase modulators represent a promising therapeutic approach for Alzheimer's disease (AD) because they selectively decrease amyloid ? 42 (A?42), a particularly neurotoxic A? species that accumulates in plaques in the brains of patients with AD. In the present study, we describe the in vitro and in vivo pharmacological properties of a potent novel ?-secretase modulator, 2-(S)-(3,5-bis(4-(trifluoromethyl)phenyl)phenyl)-4-methylpentanoic acid (JNJ-40418677). | | | 21232036
|
AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability.
Jonas, MC; Pehar, M; Puglielli, L
Journal of cell science
123
3378-88
2009
Zobrazit abstrakt
The transient or permanent modification of nascent proteins in the early secretory pathway is an essential cellular function that ensures correct folding and maturation of membrane and secreted proteins. We have recently described a new form of post-translational regulation of the membrane protein β-site APP cleaving enzyme 1 (BACE1) involving transient lysine acetylation in the lumen of the endoplasmic reticulum (ER). The essential components of this process are two ER-based acetyl-CoA:lysine acetyltransferases, ATase1 and ATase2, and a membrane transporter that translocates acetyl-CoA into the lumen of the ER. Here, we report the functional identification of acetyl-CoA transporter 1 (AT-1) as the ER membrane acetyl-CoA transporter. We show that AT-1 regulates the acetylation status of ER-transiting proteins, including the membrane proteins BACE1, low-density lipoprotein receptor and amyloid precursor protein (APP). Finally, we show that AT-1 is essential for cell viability as its downregulation results in widespread cell death and induction of features characteristic of autophagy. | | | 20826464
|
Insulin-degrading enzyme sorting in exosomes: a secretory pathway for a key brain amyloid-beta degrading protease.
Ayelén Bulloj,María C Leal,Huaxi Xu,Eduardo M Castaño,Laura Morelli
Journal of Alzheimer's disease : JAD
19
2009
Zobrazit abstrakt
The accumulation of amyloid-beta (Abeta) peptides in senile plaques is one of the hallmarks of Alzheimer's disease (AD) progression. The endocytic pathway has been proposed as a major subcellular site for Abeta generation while the compartments in which Abeta-degrading proteases interact with Abeta are still elusive. It was suggested that extracellular Abeta degradation may take place by plasma-membrane associated proteases or by extracellular proteases, among which insulin-degrading enzyme (IDE) is the most relevant. However, the mechanisms of IDE secretion are poorly understood. In the present study we used N2a cells to explore if IDE is indeed released through exosomes and the effect of exosomes release on extracellular levels of Abeta. We demonstrated that proteolytically-active plasma membrane associated-IDE is routed in living N2a cells to multivesicular bodies and subsequently, a major fraction is sorted to exosomes. We described that extracellular IDE levels decrease if the generation of multivesicular bodies is interfered and may be positively modulated by exosomes release under stress-induced conditions. Our results reinforce the relevance of functional IDE in the catabolism of extracellular Abeta. | | | 20061628
|
Zinc overload enhances APP cleavage and Aβ deposition in the Alzheimer mouse brain.
Wang, CY; Wang, T; Zheng, W; Zhao, BL; Danscher, G; Chen, YH; Wang, ZY
PloS one
5
e15349
2009
Zobrazit abstrakt
Abnormal zinc homeostasis is involved in β-amyloid (Aβ) plaque formation and, therefore, the zinc load is a contributing factor in Alzheimer's disease (AD). However, the involvement of zinc in amyloid precursor protein (APP) processing and Aβ deposition has not been well established in AD animal models in vivo.In the present study, APP and presenilin 1 (PS1) double transgenic mice were treated with a high dose of zinc (20 mg/ml ZnSO4 in drinking water). This zinc treatment increased APP expression, enhanced amyloidogenic APP cleavage and Aβ deposition, and impaired spatial learning and memory in the transgenic mice. We further examined the effects of zinc overload on APP processing in SHSY-5Y cells overexpressing human APPsw. The zinc enhancement of APP expression and cleavage was further confirmed in vitro.The present data indicate that excess zinc exposure could be a risk factor for AD pathological processes, and alteration of zinc homeostasis is a potential strategy for the prevention and treatment of AD. | | | 21179415
|