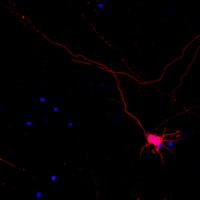
Presence of a neo-epitope and absence of amyloid beta and tau protein in degenerative hippocampal granules of aged mice.
Manich, G; del Valle, J; Cabezón, I; Camins, A; Pallàs, M; Pelegrí, C; Vilaplana, J
Age (Dordrecht, Netherlands)
36
151-65
2014
Show Abstract
Clustered pathological granules related to a degenerative process appear and increase progressively with age in the hippocampus of numerous mouse strains. We describe herein the presence of a neo-epitope of carbohydrate nature in these granules, which is not present in other brain areas and thus constitutes a new marker of these degenerative structures. We also found that this epitope is recognised by a contaminant IgM present in several antibodies obtained from mouse ascites and from both mouse and rabbit sera. These findings entail the need to revise the high number of components that are thought to be present in the granules, such as the controversial β-amyloid peptides described in the granules of senescence-accelerated mouse prone-8 (SAMP8) mice. Characterisation of the composition of SAMP8 granules, taking into account the presence of the neo-epitope and the contaminant IgM, showed that granules do not contain either β-amyloid peptides or tau protein. The presence of the neo-epitope in the granules but not in other brain areas opens up a new direction in the study of the neurodegenerative processes associated with age. The SAMP8 strain, in which the progression of the granules is enhanced, may be a useful model for this purpose. | | | 23867972
 |
Synaptic function of nicastrin in hippocampal neurons.
Lee, SH; Sharma, M; Südhof, TC; Shen, J
Proceedings of the National Academy of Sciences of the United States of America
111
8973-8
2014
Show Abstract
Synaptic dysfunction is widely thought to play a key role in the pathogenesis of Alzheimer's disease (AD). Presenilins, the major gene products involved in familial AD, are essential for short- and long-term synaptic plasticity in mature neurons as well as for the survival of cortical neurons during aging. Presenilin and nicastrin are both indispensable components of the γ-secretase complex, but it remains unknown whether presenilin regulates synaptic function in a γ-secretase-dependent or γ-secretase-independent manner and whether nicastrin plays similar roles in central synapses. In the current study, we address these questions using an electrophysiological approach to analyze nicastrin conditional knockout (cKO) mice in the hippocampal Schaffer collateral pathway. In these mice, we found that, even at 2 mo of age, deletion of nicastrin in excitatory neurons of the postnatal forebrain using Cre recombinase expressed under the control of the αCaMKII promoter led to deficits in presynaptic short-term plasticity including paired-pulse facilitation and frequency facilitation. Depletion of Ca(2+) in the endoplasmic reticulum mimics and occludes the presynaptic facilitation deficits in nicastrin cKO mice, suggesting that disrupted intracellular Ca(2+) homeostasis underlies the presynaptic deficits. In addition, NMDA receptor-mediated responses and long-term potentiation induced by theta-burst stimulation were decreased in nicastrin cKO mice at 3 mo but not at 2 mo of age. Together, these findings show that, similar to presenilins, nicastrin plays essential roles in the regulation of short- and long-term synaptic plasticity, highlighting the importance of γ-secretase in the function of mature synapses. | Western Blotting | Mouse | 24889619
|
Tau-mediated NMDA receptor impairment underlies dysfunction of a selectively vulnerable network in a mouse model of frontotemporal dementia.
Warmus, BA; Sekar, DR; McCutchen, E; Schellenberg, GD; Roberts, RC; McMahon, LL; Roberson, ED
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
16482-95
2014
Show Abstract
Frontotemporal dementia (FTD) is a neurodegenerative behavioral disorder that selectively affects the salience network, including the ventral striatum and insula. Tau mutations cause FTD, but how mutant tau impairs the salience network is unknown. Here, we address this question using a mouse model expressing the entire human tau gene with an FTD-associated mutation (V337M). Mutant, but not wild-type, human tau transgenic mice had aging-dependent repetitive and disinhibited behaviors, with synaptic deficits selectively in the ventral striatum and insula. There, mutant tau depleted PSD-95, resulting in smaller postsynaptic densities and impaired synaptic localization of NMDA receptors (NMDARs). In the ventral striatum, decreased NMDAR-mediated transmission reduced striatal neuron firing. Pharmacologically enhancing NMDAR function with the NMDAR co-agonist cycloserine reversed electrophysiological and behavioral deficits. These results indicate that NMDAR hypofunction critically contributes to FTD-associated behavioral and electrophysiological alterations and that this process can be therapeutically targeted by a Food and Drug Administration-approved drug. | | | 25471585
|
Atypical protein kinase C and Par3 are required for proteoglycan-induced axon growth inhibition.
Lee, SI; Zhang, W; Ravi, M; Weschenfelder, M; Bastmeyer, M; Levine, JM
The Journal of neuroscience : the official journal of the Society for Neuroscience
33
2541-54
2013
Show Abstract
When the CNS is injured, damaged axons do not regenerate. This failure is due in part to the growth-inhibitory environment that forms at the injury site. Myelin-associated molecules, repulsive axon guidance molecules, and extracellular matrix molecules including chondroitin sulfate proteoglycans (CSPGs) found within the glial scar inhibit axon regeneration but the intracellular signaling mechanisms triggered by these diverse molecules remain largely unknown. Here we provide biochemical and functional evidence that atypical protein kinase C (PKCζ) and polarity (Par) complex proteins mediate axon growth inhibition. Treatment of postnatal rat neurons in vitro with the NG2 CSPG, a major component of the glial scar, activates PKCζ, and this activation is both necessary and sufficient to inhibit axonal growth. NG2 treatment also activates Cdc42, increases the association of Par6 with PKCζ, and leads to a Par3-dependent activation of Rac1. Transfection of neurons with kinase-dead forms of PKCζ, dominant-negative forms of Cdc42, or mutant forms of Par6 that do not bind to Cdc42 prevent NG2-induced growth inhibition. Similarly, transfection with either a phosphomutant Par3 (S824A) or dominant-negative Rac1 prevent inhibition, whereas expression of constitutively active Rac1 inhibits axon growth on control surfaces. These results suggest a model in which NG2 binding to neurons activates PKCζ and modifies Par complex function. They also identify the Par complex as a novel therapeutic target for promoting axon regeneration after CNS injury. | | | 23392682
|
Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function.
Oaks, AW; Frankfurt, M; Finkelstein, DI; Sidhu, A
PloS one
8
e60378
2013
Show Abstract
Expression of A53T mutant human alpha-synuclein under the mouse prion promoter is among the most successful transgenic models of Parkinson's disease. Accumulation of A53T alpha-synuclein causes adult mice to develop severe motor impairment resulting in early death at 8-12 months of age. In younger, pre-symptomatic animals, altered motor activity and anxiety-like behaviors have also been reported. These behavioral changes, which precede severe neuropathology, may stem from non-pathological functions of alpha-synuclein, including modulation of monoamine neurotransmission. Our analysis over the adult life-span of motor activity, anxiety-like, and depressive-like behaviors identifies perturbations both before and after the onset of disease. Young A53T mice had increased distribution of the dopamine transporter (DAT) to the membrane that was associated with increased striatal re-uptake function. DAT function decreased with aging, and was associated with neurochemical alterations that included increased expression of beta-synuclein and gamma synuclein. Prior to normalization of dopamine uptake, transient activation of Tau kinases and hyperphosphorylation of Tau in the striatum were also observed. Aged A53T mice had reduced neuron counts in the substantia nigra pars compacta, yet striatal medium spiny neuron dendritic spine density was largely maintained. These findings highlight the involvement of the synuclein family of proteins and phosphorylation of Tau in the response to dopaminergic dysfunction of the nigrostriatal pathway. | | | 23560093
|
CSPα knockout causes neurodegeneration by impairing SNAP-25 function.
Sharma, M; Burré, J; Bronk, P; Zhang, Y; Xu, W; Südhof, TC
The EMBO journal
31
829-41
2012
Show Abstract
At a synapse, the synaptic vesicle protein cysteine-string protein-α (CSPα) functions as a co-chaperone for the SNARE protein SNAP-25. Knockout (KO) of CSPα causes fulminant neurodegeneration that is rescued by α-synuclein overexpression. The CSPα KO decreases SNAP-25 levels and impairs SNARE-complex assembly; only the latter but not the former is reversed by α-synuclein. Thus, the question arises whether the CSPα KO phenotype is due to decreased SNAP-25 function that then causes neurodegeneration, or due to the dysfunction of multiple as-yet uncharacterized CSPα targets. Here, we demonstrate that decreasing SNAP-25 levels in CSPα KO mice by either KO or knockdown of SNAP-25 aggravated their phenotype. Conversely, increasing SNAP-25 levels by overexpression rescued their phenotype. Inactive SNAP-25 mutants were unable to rescue, showing that the rescue was specific. Under all conditions, the neurodegenerative phenotype precisely correlated with SNARE-complex assembly, indicating that impaired SNARE-complex assembly due to decreased SNAP-25 levels is the ultimate correlate of neurodegeneration. Our findings suggest that the neurodegeneration in CSPα KO mice is primarily produced by defective SNAP-25 function, which causes neurodegeneration by impairing SNARE-complex assembly. | | | 22187053
|
Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways.
Wills, J; Credle, J; Oaks, AW; Duka, V; Lee, JH; Jones, J; Sidhu, A
PloS one
7
e30745
2012
Show Abstract
SNCA and MAPT genes and environmental factors are important risk factors of Parkinson's disease [PD], the second-most common neurodegenerative disease. The agrichemicals maneb and paraquat selectively target dopaminergic neurons, leading to parkinsonism, through ill-defined mechanisms. In the current studies we have analyzed the ability of maneb and paraquat, separately and together, to induce synucleinopathy and tauopathy in wild type mice. Maneb was ineffective in increasing α-synuclein [α-Syn] or p-Tau levels. By contrast, paraquat treatment of mice resulted in robust accumulation of α-Syn and hyperphosphorylation of Tau in striata, through activation of p-GSK-3β, a major Tau kinase. Co-treatment with maneb did not enhance the effects of paraquat. Increased hyperacetylation of α-tubulin was observed in paraquat-treated mice, suggesting cytoskeleton remodeling. Paraquat, but not maneb, inhibited soluble proteasomal activity on a peptide substrate but this was not associated with a decreased expression of 26S proteasome subunits. Both paraquat and maneb treatments increased levels of the autophagy inhibitor, mammalian target of rapamycin, mTOR, suggesting impaired axonal autophagy, despite increases in certain autophagic proteins, such as beclin 1 and Agt12. Autophagic flux was also impaired, as ratios of LC3 II to LC3 I were reduced in treated animals. Increased mTOR was also observed in postmortem human PD striata, where there was a reduction in the LC3 II to LC3 I ratio. Heat shock proteins were either increased or unchanged upon paraquat-treatment suggesting that chaperone-mediated autophagy is not hampered by the agrichemicals. These studies provide novel insight into the mechanisms of action of these agrichemicals, which indicate that paraquat is much more toxic than maneb, via its inhibitory effects on proteasomes and autophagy, which lead to accumulation of α-Syn and p-Tau. | | | 22292029
|
Tauopathic changes in the striatum of A53T α-synuclein mutant mouse model of Parkinson's disease.
Wills, J; Credle, J; Haggerty, T; Lee, JH; Oaks, AW; Sidhu, A
PloS one
6
e17953
2011
Show Abstract
Tauopathic pathways lead to degenerative changes in Alzheimer's disease and there is evidence that they are also involved in the neurodegenerative pathology of Parkinson's disease [PD]. We have examined tauopathic changes in striatum of the α-synuclein (α-Syn) A53T mutant mouse. Elevated levels of α-Syn were observed in striatum of the adult A53T α-Syn mice. This was accompanied by increases in hyperphosphorylated Tau [p-Tau], phosphorylated at Ser202, Ser262 and Ser396/404, which are the same toxic sites also seen in Alzheimer's disease. There was an increase in active p-GSK-3β, hyperphosphorylated at Tyr216, a major and primary kinase known to phosphorylate Tau at multiple sites. The sites of hyperphosphorylation of Tau in the A53T mutant mice were similar to those seen in post-mortem striata from PD patients, attesting to their pathophysiological relevance. Increases in p-Tau were not due to alterations on protein phosphatases in either A53T mice or in human PD, suggesting lack of involvement of these proteins in tauopathy. Extraction of striata with Triton X-100 showed large increases in oligomeric forms of α-Syn suggesting that α-Syn had formed aggregates the mutant mice. In addition, increased levels of p-GSK-3β and pSer396/404 were also found associated with aggregated α-Syn. Differential solubilization to measure protein binding to cytoskeletal proteins demonstrated that p-Tau in the A53T mutant mouse were unbound to cytoskeletal proteins, consistent with dissociation of p-Tau from the microtubules upon hyperphosphorylation. Interestingly, α-Syn remained tightly bound to the cytoskeleton, while p-GSK-3β was seen in the cytoskeleton-free fractions. Immunohistochemical studies showed that α-Syn, pSer396/404 Tau and p-GSK-3β co-localized with one another and was aggregated and accumulated into large inclusion bodies, leading to cell death of Substantia nigral neurons. Together, these data demonstrate an elevated state of tauopathy in striata of the A53T α-Syn mutant mice, suggesting that tauopathy is a common feature of synucleinopathies. Full Text Article | | | 21445308
|
Hyperphosphorylated Tau in an α-synuclein-overexpressing transgenic model of Parkinson's disease.
Haggerty, T; Credle, J; Rodriguez, O; Wills, J; Oaks, AW; Masliah, E; Sidhu, A
The European journal of neuroscience
33
1598-610
2011
Show Abstract
Although clinically distinct diseases, tauopathies and synucleinopathies share a common genesis and mechanisms, leading to overlapping degenerative changes within neurons. In human postmortem striatum of Parkinson's disease (PD) and PD with dementia, we have recently described elevated levels of tauopathy, indexed as increased hyperphosphorylated Tau (p-Tau). Here we assessed tauopathy in striatum of a transgenic animal model of PD, overexpressing human α-synuclein under the platelet-derived growth factor promoter. At 11 months of age, large and progressive increases in p-Tau in transgenic mice, hyperphosphorylated at sites reminiscent of Alzheimer's disease, were noted, along with elevated levels of α-synuclein and glycogen synthase kinase 3β phosphorylated at Tyr216 (p-GSK-3β), a major kinase involved in the hyperphosphorylation of Tau. Differential Triton X-100 extraction of striata showed the presence of aggregated α-synuclein in the transgenic mice, along with p-Tau and p-GSK-3β, which was also confirmed through immunohistochemistry. After p-Tau formation, both Tau and microtubule-associated protein 1 (MAP1) dissociated from the cytoskeleton, consistent with the diminished ability of these cytoskeleton-binding proteins to bind microtubules. Increases in free tubulin and actin were also noted, indicative of cytoskeleton remodeling and destabilization. In vivo magnetic resonance imaging of the transgenic animals showed a reduction in brain volume of transgenic mice, indicating substantial atrophy. From immunohistochemical studies, α-synuclein, p-Tau and p-GSK-3β were found to be overexpressed and co-localized in large inclusion bodies, reminiscent of Lewy bodies. The elevated state of tauopathy seen in these platelet-derived growth factor-α-synuclein mice provides further confirmation that PD may be a tauopathic disease. | | | 21453448
|
Region-specific tauopathy and synucleinopathy in brain of the alpha-synuclein overexpressing mouse model of Parkinson's disease.
Kaul, T; Credle, J; Haggerty, T; Oaks, AW; Masliah, E; Sidhu, A
BMC neuroscience
12
79
2011
Show Abstract
α-synuclein [α-Syn]-mediated activation of GSK-3β leading to increases in hyperphosphorylated Tau has been shown by us to occur in striata of Parkinson's diseased [PD] patients and in animal models of PD. In Alzheimer's disease, tauopathy exists in several brain regions; however, the pattern of distribution of tauopathy in other brain regions of PD or in animal models of PD is not known. The current studies were undertaken to analyze the distribution of tauopathy in different brain regions in a widely used mouse model of PD, the α-Syn overexpressing mouse.High levels of α-Syn levels were seen in the brain stem, with a much smaller increase in the frontal cortex; neither cerebellum nor hippocampus showed any overexpression of α-Syn. Elevated levels of p-Tau, hyperphosphorylated at Ser202, Ser262 and Ser396/404, were seen in brain stem, with lower levels seen in hippocampus. In both frontal cortex and cerebellum, increases were seen only in p-Ser396/404 Tau, but not in p-Ser202 and p-Ser262. p-GSK-3β levels were not elevated in any of the brain regions, although total GSK-3β was elevated in brain stem. p-p38MAPK levels were unchanged in all brain regions examined, while p-ERK levels were elevated in brain stem, hippocampus and cerebellum, but not the frontal cortex. p-JNK levels were increased in brain stem and cerebellum but not in the frontal cortex or hippocampus. Elevated levels of free tubulin, indicating microtubule destabilization, were seen only in the brain stem.Our combined data suggest that in this animal model of PD, tauopathy, along with microtubule destabilization, exists primarily in the brain stem and striatum, which are also the two major brain regions known to express high levels of α-Syn and undergo the highest levels of degeneration in human PD. Thus, tauopathy in PD may have a very restricted pattern of distribution. | | | 21812967
|