



04-1466 Sigma-AldrichAnti-SMN Antibody, clone 62E7
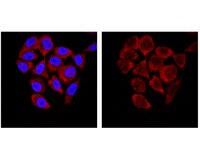


Use Anti-SMN Antibody, clone 62E7 (Mouse Monoclonal Antibody) validated in WB, ICC, IP to detect SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease).
More>> Use Anti-SMN Antibody, clone 62E7 (Mouse Monoclonal Antibody) validated in WB, ICC, IP to detect SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease). Less<<Anti-SMN Antibody, clone 62E7 MSDS (material safety data sheet) or SDS, CoA and CoQ, dossiers, brochures and other available documents.
Recommended Products
Overview
| Replacement Information |
|---|
Key Spec Table
| Species Reactivity | Key Applications | Host | Format | Antibody Type |
|---|---|---|---|---|
| H, M, Xn | WB, ICC, IP | M | Purified | Monoclonal Antibody |
| References |
|---|
| Product Information | |
|---|---|
| Format | Purified |
| Control |
|
| Presentation | Purified mouse monoclonal IgG1κ in buffer containing 0.1 M Tris-Glycine (pH 7.4, 150 mM NaCl) with 0.05% sodium azide. |
| Quality Level | MQ100 |
| Physicochemical Information |
|---|
| Dimensions |
|---|
| Materials Information |
|---|
| Toxicological Information |
|---|
| Safety Information according to GHS |
|---|
| Safety Information |
|---|
| Storage and Shipping Information | |
|---|---|
| Storage Conditions | Stable for 1 year at 2-8°C from date of receipt. |
| Packaging Information | |
|---|---|
| Material Size | 100 µg |
| Transport Information |
|---|
| Supplemental Information |
|---|
| Specifications |
|---|
| Global Trade Item Number | |
|---|---|
| Catalogue Number | GTIN |
| 04-1466 | 04053252337673 |
Documentation
Anti-SMN Antibody, clone 62E7 SDS
| Title |
|---|
Anti-SMN Antibody, clone 62E7 Certificates of Analysis
| Title | Lot Number |
|---|---|
| Anti-SMN, clone 62E7 | 2476755 |
| Anti-SMN, clone 62E7 - 2322033 | 2322033 |
| Anti-SMN, clone 62E7 - 3318424 | 3318424 |
| Anti-SMN, clone 62E7 - 3807944 | 3807944 |
| Anti-SMN, clone 62E7 - 3852301 | 3852301 |
| Anti-SMN, clone 62E7 - NG1820237 | NG1820237 |
| Anti-SMN, clone 62E7 - NG1940125 | NG1940125 |
| Anti-SMN, clone 62E7 - NRG1665141 | NRG1665141 |
| Anti-SMN, clone 62E7 -2639342 | 2639342 |
| Anti-SMN, clone 62E7 -2803229 | 2803229 |