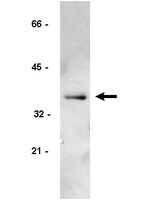
Failure of cell cleavage induces senescence in tetraploid primary cells.
Panopoulos, A; Pacios-Bras, C; Choi, J; Yenjerla, M; Sussman, MA; Fotedar, R; Margolis, RL
Molecular biology of the cell
25
3105-18
2014
Show Abstract
Tetraploidy can arise from various mitotic or cleavage defects in mammalian cells, and inheritance of multiple centrosomes induces aneuploidy when tetraploid cells continue to cycle. Arrest of the tetraploid cell cycle is therefore potentially a critical cellular control. We report here that primary rat embryo fibroblasts (REF52) and human foreskin fibroblasts become senescent in tetraploid G1 after drug- or small interfering RNA (siRNA)-induced failure of cell cleavage. In contrast, T-antigen-transformed REF52 and p53+/+ HCT116 tumor cells rapidly become aneuploid by continuing to cycle after cleavage failure. Tetraploid primary cells quickly become quiescent, as determined by loss of the Ki-67 proliferation marker and of the fluorescent ubiquitination-based cell cycle indicator/late cell cycle marker geminin. Arrest is not due to DNA damage, as the γ-H2AX DNA damage marker remains at control levels after tetraploidy induction. Arrested tetraploid cells finally become senescent, as determined by SA-β-galactosidase activity. Tetraploid arrest is dependent on p16INK4a expression, as siRNA suppression of p16INK4a bypasses tetraploid arrest, permitting primary cells to become aneuploid. We conclude that tetraploid primary cells can become senescent without DNA damage and that induction of senescence is critical to tetraploidy arrest. | Western Blotting | 25143403
 |
Serine phosphorylation regulates disabled-1 early isoform turnover independently of Reelin.
Gao, Z; Godbout, R
Cellular signalling
23
555-65
2011
Show Abstract
The Reelin-Disabled 1 (Dab1) signaling pathway plays an important role in neuronal cell migration during brain development. Dab1, an intracellular adapter protein which is tyrosine phosphorylated upon Reelin stimulation, has been directly implicated in the transmission and termination of Reelin-mediated signaling. Two main forms of Dab1 have been identified in the developing chick retina, an early isoform (Dab1-E) expressed in progenitor cells and a late isoform (Dab1-L, a.k.a. Dab1) expressed in differentiated cells. Dab1-E is missing two Src family kinase (SFK) phosphorylation sites that are critical for Reelin-Dab1 signaling and is not tyrosine phosphorylated. We have recently demonstrated a role for Dab1-E in the maintenance of retinal progenitor cells. Here, we report that Dab1-E is phosphorylated at serine/threonine residues independent of Reelin. Cdk2, highly expressed in retinal progenitor cells, mediates Dab1-E phosphorylation at serine 475 which in turn promotes ubiquitination-triggered proteasome degradation of Dab1-E. Inhibition of protein phosphatase 1 and/or protein phosphatase 2A leads to increased Dab1-E instability. We propose that Dab1 turnover is regulated by both Reelin-independent serine/threonine phosphorylation and Reelin-dependent tyrosine phosphorylation. | Western Blotting | 21111810
|
Cooperative signaling between Wnt1 and integrin-linked kinase induces accelerated breast tumor development.
Oloumi, A; Maidan, M; Lock, FE; Tearle, H; McKinney, S; Muller, WJ; Aparicio, SA; Dedhar, S
Breast cancer research : BCR
12
R38
2010
Show Abstract
Breast cancer is genetically and clinically a heterogeneous disease. However, the exact contribution of different cell types and oncogenic mutations to this heterogeneity are not well understood. Recently, we discovered an interaction between Wnt and integrin-linked kinase (ILK) within the signaling cascade that regulates cell growth and survival. Interestingly, mammary-specific expression of either one of these proteins has been shown to promote mammary tumorigenesis. In light of our recent findings and to investigate the potential interaction between Wnt and ILK proteins during mammary tumor formation and progression, we established a transgenic mouse model that expresses both Wnt and ILK in mammary epithelial cells.A novel transgenic mouse model with mammary-specific expression of both Wnt1 and ILK was generated by crossing the two previously characterized mouse models, MMTV-Wnt1 and MMTV-ILK. The resulting MMTV-Wnt/ILK mice were closely monitored for tumor development and growth, as well as for the tumor onset. The molecular phenotypes of both tumors and premalignant mammary glands were investigated by using biochemical and global gene-expression analysis approaches.A significant acceleration in mammary tumor incidence and growth was observed in the MMTV-Wnt/ILK mice. Pre-neoplastic mammary glands also display lobuloalveolar hyperplasia and an increase in ductal epithelium proliferation. Apart from elevated expression of Wnt/ILK targets, such as beta-catenin and cyclin D1, gene-expression profiling identified the surprising activation of the FOXA1 transcription factor. Upregulation of FOXA1, which is also known as the molecular marker of differentiated mammary luminal cells, was consistent with the expansion of the enriched luminal progenitor population or CD29loCD24hiCD61+ cells in MMTV-Wnt/ILK tumors.These results show cooperation between Wnt1 and ILK transgenes during mammary carcinogenesis, leading to changes in a transcriptional network, which could dictate a specific breast cancer phenotype with enhanced growth dynamics. The MMTV-Wnt/ILK can be used as a model to identify further the genes downstream of the estrogen receptor-beta/FOXA1 and to investigate the mechanisms targeting the expansion of the luminal progenitor cells leading to hyperplasia and tumorigenesis. | Western Blotting | 20565980
|
Akt2-mediated phosphorylation of Pitx2 controls Ccnd1 mRNA decay during muscle cell differentiation.
R Gherzi,M Trabucchi,M Ponassi,I-E Gallouzi,M G Rosenfeld,P Briata
Cell death and differentiation
17
2010
Show Abstract
Paired-like homeodomain 2 (Pitx2), first identified as the gene responsible for the Axenfeld-Rieger syndrome, encodes a protein factor that, controlling cell proliferation in a tissue-specific manner, has a crucial role in morphogenesis. During embryonic development, Pitx2 exerts a role in the expansion of muscle progenitors and is expressed at all stages of myogenic progression. In this study, we show that Pitx2 is phosphorylated by the protein kinase Akt2 and is necessary to ensure proper C2C12 myoblast proliferation and differentiation. Pitx2 associates with a ribonucleoprotein complex that includes the mRNA stabilizing factor HuR and sustains Ccnd1 (also known as Cyclin D1) expression, thereby prolonging its mRNA half-life. When the differentiation program is initiated, phosphorylation by Akt2 impairs the ability of Pitx2 to associate with the Ccnd1 mRNA-stabilizing complex that includes HuR and, as a consequence, Ccnd1 mRNA half-life is shortened. We propose that unphosphorylated Pitx2 is required to favor HuR-mediated Ccnd1 mRNA stabilization, thus sustaining myoblast proliferation. Upon Akt2-phosphorylation, the complex Pitx2/HuR/Ccnd1 mRNA dissociates and Ccnd1 mRNA is destabilized. These events contribute to the switch of C2C12 cells from a proliferating to a differentiating phenotype. | | 20019746
|
INK4a/Arf is required for suppression of EGFR/DeltaEGFR(2-7)-dependent ERK activation in mouse astrocytes and glioma.
Lachat, Y; Diserens, AC; Nozaki, M; Kobayashi, H; Hamou, MF; Godard, S; De Tribolet, N; Hegi, ME
Oncogene
23
6854-63
2004
Show Abstract
Amplification of the epidermal growth factor receptor (EGFR) or expression of its constitutively activated mutant, DeltaEGFR(2-7), in association with the inactivation of the INK4a/Arf gene locus is a frequent alteration in human glioblastoma. The notion of a cooperative effect between these two alterations has been demonstrated in respective mouse brain tumor models including our own. Here, we investigated underlying molecular mechanisms in early passage cortical astrocytes deficient for p16(INK4a)/p19(Arf) or p53, respectively, with or without ectopic expression of DeltaEGFR(2-7). Targeting these cells with the specific EGFR inhibitor tyrphostin AG1478 revealed that phosphorylation of ERK was only abrogated in the presence of an intact INK4a/Arf gene locus. The sensitivity to inhibit ERK phosphorylation was independent of ectopic expression of DeltaEGFR(2-7) and independent of the TP53 status. This resistance to downregulate the MAPK pathway in the absence of INK4a/Arf was confirmed in cell lines derived from our mouse glioma models with the respective initial genetic alterations. Thus, deletion of INK4a/Arf appears to keep ERK in its active, phosphorylated state insensitive to an upstream inhibitor specifically targeting EGFR/DeltaEGFR(2-7). This resistance may contribute to the cooperative tumorigenic effect selected for in human glioblastoma that may be of crucial clinical relevance for treatments specifically targeting EGFR/DeltaEGFR(2-7) in glioblastoma patients. | Immunohistochemistry | 15273738
|
A non-thiazolidinedione partial peroxisome proliferator-activated receptor gamma ligand inhibits vascular smooth muscle cell growth.
Dennis Bruemmer, Joel P Berger, Joey Liu, Ulrich Kintscher, Shu Wakino, Eckart Fleck, David E Moller, Ronald E Law, Dennis Bruemmer, Joel P Berger, Joey Liu, Ulrich Kintscher, Shu Wakino, Eckart Fleck, David E Moller, Ronald E Law
European journal of pharmacology
466
225-34
2003
Show Abstract
Several peroxisome proliferator-activated receptor gamma (PPARgamma) agonists of the thiazolidinedione class inhibit vascular smooth muscle cell proliferation. It is not known whether the antiproliferative activity of PPARgamma agonists is limited to the thiazolidinedione class and/or is directly mediated through PPARgamma-dependent transactivation of target genes. We report here that a novel non-thiazolidinedione partial PPARgamma agonist (nTZDpa) attenuates rat aortic vascular smooth muscle cell proliferation. In a transfection assay for PPARgamma transcriptional activation, the non-thiazolidinedione partial PPARgamma agonist elicited approximately 25% of the maximal efficacy of the full PPARgamma agonist rosiglitazone. In the presence of the non-thiazolidinedione partial PPARgamma agonist, the transcriptional activity of the full agonist, rosiglitazone, was blunted, indicating that the non-thiazolidinedione partial PPARgamma agonist inhibits rosiglitazone-induced PPARgamma activity. The non-thiazolidinedione partial PPARgamma agonist (0.1-10 microM) inhibited vascular smooth muscle cell growth which was accompanied by an inhibition of retinoblastoma protein phosphorylation. Mitogen-induced downregulation of the cyclin-dependent kinase (CDK) inhibitor p27(kip1), and induction of the G1 cyclins cyclin D1, cyclin A, and cyclin E were also attenuated by the non-thiazolidinedione partial PPARgamma agonist. Maximal antiproliferative activity of the non-thiazolidinedione partial PPARgamma agonist required functional PPARgamma as adenovirus-mediated overexpression of a dominant-negative PPARgamma mutant partially reversed its inhibition of vascular smooth muscle cell growth. In contrast, overexpression of dominant-negative PPARgamma did not reverse the inhibitory effect of the non-thiazolidinedione partial PPARgamma agonist on cyclin D1. As the full PPARgamma agonist rosiglitazone exhibited no effect on cyclin D1, inhibition of that G1 cyclin by the non-thiazolidinedione partial PPARgamma agonist likely occurred through a PPARgamma-independent mechanism. These data demonstrate that a non-thiazolidinedione partial PPARgamma agonist may constitute a novel therapeutic for proliferative vascular diseases and could provide additional evidence for the important role of PPARgamma in regulating vascular smooth muscle cell proliferation. | | 12694805
|
Tumor suppressor PTEN inhibits nuclear accumulation of beta-catenin and T cell/lymphoid enhancer factor 1-mediated transcriptional activation.
Persad, S, et al.
J. Cell Biol., 153: 1161-74 (2001)
2001
Show Abstract
beta-Catenin is a protein that plays a role in intercellular adhesion as well as in the regulation of gene expression. The latter role of beta-catenin is associated with its oncogenic properties due to the loss of expression or inactivation of the tumor suppressor adenomatous polyposis coli (APC) or mutations in beta-catenin itself. We now demonstrate that another tumor suppressor, PTEN, is also involved in the regulation of nuclear beta-catenin accumulation and T cell factor (TCF) transcriptional activation in an APC-independent manner. We show that nuclear beta-catenin expression is constitutively elevated in PTEN null cells and this elevated expression is reduced upon reexpression of PTEN. TCF promoter/luciferase reporter assays and gel mobility shift analysis demonstrate that PTEN also suppresses TCF transcriptional activity. Furthermore, the constitutively elevated expression of cyclin D1, a beta-catenin/TCF-regulated gene, is also suppressed upon reexpression of PTEN. Mechanistically, PTEN increases the phosphorylation of beta-catenin and enhances its rate of degradation. We define a pathway that involves mainly integrin-linked kinase and glycogen synthase kinase 3 in the PTEN-dependent regulation of beta-catenin stability, nuclear beta-catenin expression, and transcriptional activity. Our data indicate that beta-catenin/TCF-mediated gene transcription is regulated by PTEN, and this may represent a key mechanism by which PTEN suppresses tumor progression. | Immunoblotting (Western) | 11402061
|
Dual stimulation of Ras/mitogen-activated protein kinase and RhoA by cell adhesion to fibronectin supports growth factor-stimulated cell cycle progression
Danen, E. H., et al
J Cell Biol, 151:1413-22 (2000)
2000
| Immunoblotting (Western) | 11134071
|
NA22598, a novel antitumor compound, reduces cyclin D1 levels, arrests cell cycle at G1 phase, and inhibits anchorage-independent growth of human tumor cells.
M Kawada, A Kuwahara, T Nishikiori, S Mizuno, Y Uehara
Experimental cell research
249
240-7
1999
Show Abstract
NA22598, a novel antitumor compound isolated from a microbial cultured broth, inhibited the growth of human colon cancer DLD-1 cells in suspension cultures (anchorage-independent growth) severalfold more strongly than in substratum-attached monolayer cultures. It arrested the cell cycle progression at early G1 phase under both these culture conditions. Rb phosphorylation, cyclin D1 expression, and cdk2 activation in G1 progression were all inhibited by NA22598, but the amounts of cdk2 and p27 were not affected. Among these effects the inhibition of cyclin D1 expression was most prominent, and NA22598 was found to inhibit the synthesis of cyclin D1 without affecting mRNA expression or protein degradation. p27 binding to cdk2 was more markedly increased in suspension cultures than in attached cultures by NA22598, but the compound had no effect on total p27. Apparently, the decrease of cyclin D1 induced redistribution of p27 from the cyclin D1/cdk4 to the cyclin E/cdk2 complexes during G1 phase in the suspension cultures. Because p27 is upregulated during suspension culture, a greater amount of it was associated with cyclin E/cdk2, thus producing greater growth inhibition. An agent, like NA22598, which induces the downregulation of cyclin D1 might offer a new anticancer strategy. | | 10366423
|
Monoclonal antibody against PRAD1/cyclin D1 stains nuclei of tumor cells with translocation or amplification at BCL-1 locus.
Banno, S, et al.
Jpn. J. Cancer Res., 85: 918-26 (1994)
1994
| | 7961120
|