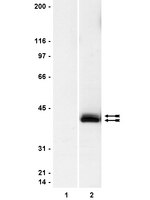
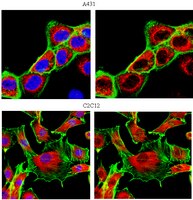
Characterization of Aldh2 (-/-) mice as an age-related model of cognitive impairment and Alzheimer's disease.
D'Souza, Y; Elharram, A; Soon-Shiong, R; Andrew, RD; Bennett, BM
Molecular brain
8
27
2015
Pokaż streszczenie
The study of late-onset/age-related Alzheimer's disease (AD)(sporadic AD, 95% of AD cases) has been hampered by a paucity of animal models. Oxidative stress is considered a causative factor in late onset/age-related AD, and aldehyde dehydrogenase 2 (ALDH2) is important for the catabolism of toxic aldehydes associated with oxidative stress. One such toxic aldehyde, the lipid peroxidation product 4-hydroxynonenal (HNE), accumulates in AD brain and is associated with AD pathology. Given this linkage, we hypothesized that in mice lacking ALDH2, there would be increases in HNE and the appearance of AD-like pathological changes.Changes in relevant AD markers in Aldh2 (-/-) mice and their wildtype littermates were assessed over a 1 year period. Marked increases in HNE adducts arise in hippocampi from Aldh2 (-/-) mice, as well as age-related increases in amyloid-beta, p-tau, and activated caspases. Also observed were age-related decreases in pGSK3β, PSD95, synaptophysin, CREB and pCREB. Age-related memory deficits in the novel object recognition and Y maze tasks begin at 3.5-4 months and are maximal at 6.5-7 months. There was decreased performance in the Morris Water Maze task in 6 month old Aldh2 (-/-) mice. These mice exhibited endothelial dysfunction, increased amyloid-beta in cerebral microvessels, decreases in carbachol-induced pCREB and pERK formation in hippocampal slices, and brain atrophy. These AD-associated pathological changes are rarely observed as a constellation in current AD animal models.We believe that this new model of age-related cognitive impairment will provide new insight into the pathogenesis and molecular/cellular mechanisms driving neurodegenerative diseases of aging such as AD, and will prove useful for assessing the efficacy of therapeutic agents for improving memory and for slowing, preventing, or reversing AD progression. | | 25910195
 |
MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition.
Taylor, DM; Moser, R; Régulier, E; Breuillaud, L; Dixon, M; Beesen, AA; Elliston, L; Silva Santos, Mde F; Kim, J; Jones, L; Goldstein, DR; Ferrante, RJ; Luthi-Carter, R
The Journal of neuroscience : the official journal of the Society for Neuroscience
33
2313-25
2013
Pokaż streszczenie
We previously demonstrated that sodium butyrate is neuroprotective in Huntington's disease (HD) mice and that this therapeutic effect is associated with increased expression of mitogen-activated protein kinase/dual-specificity phosphatase 1 (MKP-1/DUSP1). Here we show that enhancing MKP-1 expression is sufficient to achieve neuroprotection in lentiviral models of HD. Wild-type MKP-1 overexpression inhibited apoptosis in primary striatal neurons exposed to an N-terminal fragment of polyglutamine-expanded huntingtin (Htt171-82Q), blocking caspase-3 activation and significantly reducing neuronal cell death. This neuroprotective effect of MKP-1 was demonstrated to be dependent on its enzymatic activity, being ablated by mutation of its phosphatase domain and being attributed to inhibition of specific MAP kinases (MAPKs). Overexpression of MKP-1 prevented the polyglutamine-expanded huntingtin-induced activation of c-Jun N-terminal kinases (JNKs) and p38 MAPKs, whereas extracellular signal-regulated kinase (ERK) 1/2 activation was not altered by either polyglutamine-expanded Htt or MKP-1. Moreover, mutants of MKP-1 that selectively prevented p38 or JNK binding confirmed the important dual contributions of p38 and JNK regulation to MKP-1-mediated neuroprotection. These results demonstrate additive effects of p38 and JNK MAPK inhibition by MKP-1 without consequence to ERK activation in this striatal neuron-based paradigm. MKP-1 also provided neuroprotection in vivo in a lentiviral model of HD neuropathology in rat striatum. Together, these data extend previous evidence that JNK- and p38-mediated pathways contribute to HD pathogenesis and, importantly, show that therapies simultaneously inhibiting both JNK and p38 signaling pathways may lead to improved neuroprotective outcomes. | Western Blotting, Immunohistochemistry, Immunocytochemistry | 23392662
|
Decreased striatal RGS2 expression is neuroprotective in Huntington's disease (HD) and exemplifies a compensatory aspect of HD-induced gene regulation.
Seredenina, T; Gokce, O; Luthi-Carter, R
PloS one
6
e22231
2010
Pokaż streszczenie
The molecular phenotype of Huntington's disease (HD) is known to comprise highly reproducible changes in gene expression involving striatal signaling genes. Here we test whether individual changes in striatal gene expression are capable of mitigating HD-related neurotoxicity.We used protein-encoding and shRNA-expressing lentiviral vectors to evaluate the effects of RGS2, RASD2, STEP and NNAT downregulation in HD. Of these four genes, only RGS2 and RASD2 modified mutant htt fragment toxicity in cultured rat primary striatal neurons. In both cases, disease modulation was in the opposite of the predicted direction: whereas decreased expression of RGS2 and RASD2 was associated with the HD condition, restoring expression enhanced degeneration of striatal cells. Conversely, silencing of RGS2 or RASD2 enhanced disease-related changes in gene expression and resulted in significant neuroprotection. These results indicate that RGS2 and RASD2 downregulation comprises a compensatory response that allows neurons to better tolerate huntingtin toxicity. Assessment of the possible mechanism of RGS2-mediated neuroprotection showed that RGS2 downregulation enhanced ERK activation. These results establish a novel link between the inhibition of RGS2 and neuroprotective modulation of ERK activity.Our findings both identify RGS2 downregulation as a novel compensatory response in HD neurons and suggest that RGS2 inhibition might be considered as an innovative target for neuroprotective drug development. | Immunofluorescence | 21779398
|
The 5-HT 2A serotonin receptor enhances cell viability, affects cell cycle progression and activates MEK-ERK1/2 and JAK2-STAT3 signalling pathways in human choriocarcinoma cell lines.
Oufkir T, Arseneault M, Sanderson JT, Vaillancourt C
Placenta
31
439-47. Epub 2010 Mar 25.
2009
Pokaż streszczenie
Previous results from our group have demonstrated the expression of the 5-HT(2A) receptor and a mitogenic effect of serotonin in human trophoblast. The objectives of the present study were to investigate the role of the 5-HT(2A) receptor in trophoblast cells and to determine the signalling pathways activated by this receptor. We investigated the effect of (+/-)-2,5-dimethoxy-4-iodoamphetamine hydrochloride (DOI), a selective 5-HT(2A) agonist, on cell cycle progression and cell viability in BeWo and JEG-3 cells. We also investigated, by co-immunoprecipitation and western blot analysis, the involvement of the MEK-ERK1/2 and JAK2-STAT3 signalling pathways following activation of the placental 5-HT(2A) receptor. Our results showed a concentration-dependent increase of cell viability by DOI, which was reversed by ketanserin, a selective 5-HT(2A) receptor antagonist. Furthermore, activation of the 5-HT(2A) receptor by DOI increased cell entry into the G2/M and S phase (DNA synthesis) in BeWo and JEG-3 cells, respectively. In addition, stimulation of BeWo and JEG-3 cells by DOI activated both the MEK-ERK1/2 and the JAK2-STAT3 signalling pathways. This study demonstrated that the 5-HT(2A) receptor increases cell viability and affects cell cycle progression in human trophoblast cell lines as well as activates the MEK-ERK1/2 and JAK2-STAT3 intracellular signalling pathways, which are related to survival, differentiation, migration and invasion. These findings indicate that serotonin through the activation of the 5-HT(2A) receptor is a key regulator of placentation and may play a role in the pathophysiology of certain pregnancy disorders associated with alterations in placental development, such as preeclampsia, gestational diabetes and preterm birth. | | 20338635
|
The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth.
Dormoy, V; Danilin, S; Lindner, V; Thomas, L; Rothhut, S; Coquard, C; Helwig, JJ; Jacqmin, D; Lang, H; Massfelder, T
Molecular cancer
8
123
2009
Pokaż streszczenie
Human clear cell renal cell carcinoma (CRCC) remains resistant to therapies. Recent advances in Hypoxia Inducible Factors (HIF) molecular network led to targeted therapies, but unfortunately with only limited clinical significance. Elucidating the molecular processes involved in kidney tumorigenesis and resistance is central to the development of improved therapies, not only for kidney cancer but for many, if not all, cancer types. The oncogenic PI3K/Akt, NF-kB and MAPK pathways are critical for tumorigenesis. The sonic hedgehog (SHH) signaling pathway is crucial to normal development.By quantitative RT-PCR and immunoblot, we report that the SHH signaling pathway is constitutively reactivated in tumors independently of the von Hippel-Lindau (VHL) tumor suppressor gene expression which is inactivated in the majority of CRCC. The inhibition of the SHH signaling pathway by the specific inhibitor cyclopamine abolished CRCC cell growth as assessed by cell counting, BrdU incorporation studies, fluorescence-activated cell sorting and beta-galactosidase staining. Importantly, inhibition of the SHH pathway induced tumor regression in nude mice through inhibition of cell proliferation and neo-vascularization, and induction of apoptosis but not senescence assessed by in vivo studies, immunoblot and immunohistochemistry. Gli1, cyclin D1, Pax2, Lim1, VEGF, and TGF-beta were exclusively expressed in tumors and were shown to be regulated by SHH, as evidenced by immunoblot after SHH inhibition. Using specific inhibitors and immunoblot, the activation of the oncogenic PI3K/Akt, NF-kB and MAPK pathways was decreased by SHH inhibition.These findings support targeting SHH for the treatment of CRCC and pave the way for innovative and additional investigations in a broad range of cancers. | Western Blotting | 20015350
|
Decoy receptor 3, upregulated by Epstein-Barr virus latent membrane protein 1, enhances nasopharyngeal carcinoma cell migration and invasion.
Ho, CH; Chen, CL; Li, WY; Chen, CJ
Carcinogenesis
30
1443-51
2009
Pokaż streszczenie
Decoy receptor 3 (DcR3), a member of tumor necrosis factor receptor superfamily, has been implicated in tumorigenesis through its abilities to modulate immune responses and induce angiogenesis. Epstein-Barr virus (EBV), a ubiquitous gamma-herpesvirus, is associated with malignancies including nasopharyngeal carcinoma (NPC). Previous studies show that DcR3 is overexpressed in EBV-positive lymphomas and Rta, an EBV transcription activator, can upregulate DcR3 in Burkitt lymphoma cell lines. However, DcR3 expression has not been demonstrated in EBV-associated NPC nor have there been any EBV latent genes linked to DcR3 upregulation. Here, we showed DcR3 was overexpressed in NPC. Higher DcR3 expression score and DcR3-positive rate were found in metastatic NPC than in primary NPC tissues, suggesting DcR3 may enhance cell metastatic potential. This hypothesis is supported by our observation that NPC HONE-1 cells overexpressing DcR3 exhibited significant higher migration and invasion abilities in vitro. We found besides Rta, EBV latent membrane protein (LMP) 1 can upregulate DcR3 via nuclear factor-kappaB and phosphatidylinositol 3-kinase-signaling events. Approximate 75% of LMP1-positive NPC tissues overexpressed DcR3, suggesting LMP1 may enhance DcR3 expression in vivo. Data herein suggested that increasing DcR3 expression by LMP1 not only helps EBV-associated cancer cells gain survival advantage by preventing host immune detection but also increases the chance of cancer metastasis by enhancing cell migration and invasion. All these DcR3-mediated events facilitate normal cells to gain cancer hallmarks. | | 19483191
|
Preconditioning-mimetics bradykinin and DADLE activate PI3-kinase through divergent pathways.
Michael V Cohen, Sebastian Philipp, Thomas Krieg, Lin Cui, Atsushi Kuno, Viktoriya Solodushko, James M Downey, Michael V Cohen, Sebastian Philipp, Thomas Krieg, Lin Cui, Atsushi Kuno, Viktoriya Solodushko, James M Downey
Journal of molecular and cellular cardiology
42
842-51
2007
Pokaż streszczenie
We previously reported that pharmacological preconditioning of rabbit hearts with acetylcholine involves activation of phosphatidylinositol 3-kinase (PI3-K) through transactivation of the epidermal growth factor receptor (EGFR). Transactivation is thought to be initiated by cleavage of membrane-bound pro-heparin-binding EGF-like growth factor (HB-EGF) by a membrane metalloproteinase thus releasing HB-EGF which binds to the EGFR. This pathway leads to redox signaling with the generation of reactive oxygen species (ROS) by mitochondria. We tested whether preconditioning's physiological triggers, bradykinin and opioid, also signal through the EGFR. Both bradykinin and the synthetic delta-opioid agonist DADLE increased ROS production in isolated cardiomyocytes by approximately 50%. DADLE's effect was abrogated by either metalloproteinase inhibitor III (MPI) or the diphtheria toxin mutant CRM-197 which blocks heparin-binding EGF shedding indicating that DADLE signals through EGFR transactivation. MPI also blocked DADLE's infarct-sparing effect in whole hearts. Additionally, blocking Src kinase (a component of the EGFR's signaling complex) with PP2 or PI3-K with wortmannin blocked DADLE's effect on cardiomyocyte ROS production and PP2 blocked DADLE's salvage of ischemic myocardium. Finally, DADLE increased phosphorylation of Akt and extracellular signal-regulated protein kinases (ERK) 1/2 in left ventricular myocardium, and this increase was blocked by the EGFR antagonist AG1478. On the other hand, neither MPI nor CRM-197 prevented bradykinin from increasing ROS production, and MPI did not affect bradykinin's infarct-sparing effect in intact hearts. Conversely, both PP2 and wortmannin blocked bradykinin's effect on ROS generation and also aborted bradykinin's cardioprotective effect in intact hearts. While bradykinin also increased phosphorylation of Akt and ERK in myocardium, that increase was not affected by AG1478. Hence bradykinin, unlike acetylcholine or opioid, does not transactivate EGFR, although all 3 agonists do signal through Src and PI3-K. Pełny tekst artykułu | | 17292392
|
Serum- and glucocorticoid-inducible kinase (SGK) is a target of the MAPK/ERK signaling pathway that mediates memory formation in rats
Lee, Ching-Tien, et al
Eur J Neurosci, 23:1311-20 (2006)
2005
| | 16553792
|
Localizing extracellular signal-regulated kinase (ERK) in pharmacological preconditioning's trigger pathway.
Sebastian Philipp, Stuart D Critz, Lin Cui, Viktoriya Solodushko, Michael V Cohen, James M Downey
Basic research in cardiology
101
159-67
2005
Pokaż streszczenie
Acetylcholine (ACh) and opioid receptor agonists trigger the preconditioned phenotype through sequential activation of the epidermal growth factor (EGF) receptor, phosphatidylinositol 3-kinase (PI3-K), Akt, and nitric oxide synthase (NOS), and opening of mitochondrial (mito) K(ATP) channels with the generation of reactive oxygen species (ROS). Although extracellular signal-regulated kinase (ERK) has recently been reported to be part of this pathway, its location has not been determined. To address this issue, we administered a 5-min pulse of ACh (550 microM) prior to 30 min of ischemia in isolated rabbit hearts. It reduced infarction from 30.4 +/- 2.2% of the risk zone in control hearts to 12.3 +/- 2.8% and co-administration of the MEK, and, therefore, downstream ERK inhibitor U0126 abolished protection (29.1 +/- 4.6% infarction) con.rming ERK's involvement. MitoK(ATP) opening was monitored in adult rabbit cardiomyocytes by measuring ROS production with MitoTracker Red. ROS production was increased by each of three G protein-coupled agonists: ACh (250 microM), bradykinin (BK) (500 nM), and the delta-opioid agonist DADLE (20 nM). Co-incubation with the MEK inhibitors U0126 (500 nM) or PD 98059 (10 microM) blocked the increased ROS production seen with all three agonists. Direct activation of its receptor by EGF increased ROS production and PD 98059 blocked that increase, thus placing ERK downstream of the EGF receptor. Desferoxamine (DFO) which opens mitoK(ATP) through direct activation of NOS also increased ROS. PD 98059 could not block DFO-induced ROS production, placing ERK upstream of NOS. In isolated hearts, ACh caused phosphorylation of both Akt and ERK. U0126 blocked phosphorylation of ERK but not of Akt. The PI3-K inhibitor wortmannin blocked both. Together these data indicate that ERK is located between Akt and NOS. | | 16283591
|
Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer.
Wang, SJ; Bourguignon, LY
Archives of otolaryngology--head & neck surgery
132
771-8
2005
Pokaż streszczenie
To investigate whether hyaluronan (HA) and CD44 (hereinafter HA-CD44) promotes head and neck squamous cell carcinoma (HNSCC) chemotherapy resistance and whether HA-CD44 promotes epidermal growth factor receptor (EGFR)-mediated oncogenic signaling to alter chemotherapy sensitivity in HNSCC. Hyaluronan, a glycosaminoglycan component of the extracellular matrix, is a ligand for the transmembrane receptor CD44, which acts through multiple signaling pathways to influence cellular behavior. We recently determined that HA-CD44 promotes phospholipase C-mediated calcium signaling and cisplatin resistance in HNSCC.Cell line study.Tumor cell growth with various chemotherapeutic drugs (methotrexate, doxorubicin hydrochloride, adriamycin, and cisplatin) was measured in the presence or absence of HA and other inhibitors of the EGFR-mediated signaling pathway. Immunoblotting was used to study EGFR signaling. Migration assays provided one measure of tumor progression.The addition of HA, but not HA plus anti-CD44 antibody, resulted in a 2-fold reduced ability of methotrexate and an 8-fold reduced ability of adriamycin to cause HNSCC cell death. Immunoblotting studies demonstrated that HA can promote an association between CD44 and EGFR as well as CD44-dependent activation of EGFR-mediated signaling. Migration assays demonstrated that HA-CD44 can promote tumor migration with EGFR signaling. The presence of AG1478, an EGFR inhibitor, and U0126, an extracellular signal-regulated kinase inhibitor, inhibited HA-mediated tumor growth, migration, and chemotherapy resistance.Our results indicate that HA promotes CD44/EGFR interaction, EGFR-mediated oncogenic signaling, and chemotherapy resistance in HNSCC. Perturbation of HA-CD44-mediated signaling may be a promising and novel strategy to treat HNSCC. | | 16847188
|