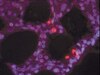
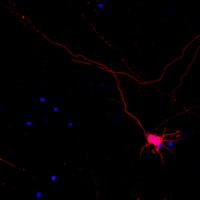
Saccadic Palsy following Cardiac Surgery: Possible Role of Perineuronal Nets.
Eggers, SD; Horn, AK; Roeber, S; Härtig, W; Nair, G; Reich, DS; Leigh, RJ
PloS one
10
e0132075
2015
Show Abstract
Perineuronal nets (PN) form a specialized extracellular matrix around certain highly active neurons within the central nervous system and may help to stabilize synaptic contacts, promote local ion homeostasis, or play a protective role. Within the ocular motor system, excitatory burst neurons and omnipause neurons are highly active cells that generate rapid eye movements - saccades; both groups of neurons contain the calcium-binding protein parvalbumin and are ensheathed by PN. Experimental lesions of excitatory burst neurons and omnipause neurons cause slowing or complete loss of saccades. Selective palsy of saccades in humans is reported following cardiac surgery, but such cases have shown normal brainstem neuroimaging, with only one clinicopathological study that demonstrated paramedian pontine infarction. Our objective was to test the hypothesis that lesions of PN surrounding these brainstem saccade-related neurons may cause saccadic palsy.Together with four controls we studied the brain of a patient who had developed a permanent selective saccadic palsy following cardiac surgery and died several years later. Sections of formalin-fixed paraffin-embedded brainstem blocks were applied to double-immunoperoxidase staining of parvalbumin and three different components of PN. Triple immunofluorescence labeling for all PN components served as internal controls. Combined immunostaining of parvalbumin and synaptophysin revealed the presence of synapses.Excitatory burst neurons and omnipause neurons were preserved and still received synaptic input, but their surrounding PN showed severe loss or fragmentation.Our findings support current models and experimental studies of the brainstem saccade-generating neurons and indicate that damage to PN may permanently impair the function of these neurons that the PN ensheathe. How a postulated hypoxic mechanism could selectively damage the PN remains unclear. We propose that the well-studied saccadic eye movement system provides an accessible model to evaluate the role of PN in health and disease. | | | 26135580
 |
Active and passive immunization strategies based on the SDPM1 peptide demonstrate pre-clinical efficacy in the APPswePSEN1dE9 mouse model for Alzheimer's disease.
Camboni, M; Wang, CM; Miranda, C; Yoon, JH; Xu, R; Zygmunt, D; Kaspar, BK; Martin, PT
Neurobiology of disease
62
31-43
2014
Show Abstract
Recent clinical and pre-clinical studies suggest that both active and passive immunization strategies targeting Aβ amyloid may have clinical benefit in Alzheimer's disease. Here, we demonstrate that vaccination of APPswePSEN1dE9 mice with SDPM1, an engineered non-native Aβ amyloid-specific binding peptide, lowers brain Aβ amyloid plaque burden and brain Aβ1-40 and Aβ1-42 peptide levels, improves cognitive learning and memory in Morris water maze tests and increases the expression of synaptic brain proteins. This was the case in young mice immunized prior to development of significant brain amyloid burden, and in older mice, where brain amyloid was already present. Active immunization was optimized using ALUM as an adjuvant to stimulate production of anti-SDPM1 and anti-Aβ amyloid antibodies. Intracerebral injection of P4D6, an SDPM1 peptide-mimotope antibody, also lowered brain amyloid plaque burden in APPswePSEN1dE9 mice. Additionally, P4D6 inhibited Aβ amyloid-mediated toxicity in cultured neuronal cells. The protein sequence of the variable domain within the P4D6 heavy chain was found to mimic a multimer of the SDPM1 peptide motif. These data demonstrate the efficacy of active and passive vaccine strategies to target Aβ amyloid oligomers using an engineered peptide-mimotope strategy. | Western Blotting | Mouse | 24021662
|
Genetic modulation of soluble Aβ rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease.
Fowler, SW; Chiang, AC; Savjani, RR; Larson, ME; Sherman, MA; Schuler, DR; Cirrito, JR; Lesné, SE; Jankowsky, JL
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
7871-85
2014
Show Abstract
An unresolved debate in Alzheimer's disease (AD) is whether amyloid plaques are pathogenic, causing overt physical disruption of neural circuits, or protective, sequestering soluble forms of amyloid-β (Aβ) that initiate synaptic damage and cognitive decline. Few animal models of AD have been capable of isolating the relative contribution made by soluble and insoluble forms of Aβ to the behavioral symptoms and biochemical consequences of the disease. Here we use a controllable transgenic mouse model expressing a mutant form of amyloid precursor protein (APP) to distinguish the impact of soluble Aβ from that of deposited amyloid on cognitive function and synaptic structure. Rapid inhibition of transgenic APP modulated the production of Aβ without affecting pre-existing amyloid deposits and restored cognitive performance to the level of healthy controls in Morris water maze, radial arm water maze, and fear conditioning. Selective reduction of Aβ with a γ-secretase inhibitor provided similar improvement, suggesting that transgene suppression restored cognition, at least in part by lowering Aβ. Cognitive improvement coincided with reduced levels of synaptotoxic Aβ oligomers, greater synaptic density surrounding amyloid plaques, and increased expression of presynaptic and postsynaptic markers. Together these findings indicate that transient Aβ species underlie much of the cognitive and synaptic deficits observed in this model and demonstrate that significant functional and structural recovery can be attained without removing deposited amyloid. | Western Blotting | | 24899710
|
The Alzheimer's β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques.
Kandalepas, PC; Sadleir, KR; Eimer, WA; Zhao, J; Nicholson, DA; Vassar, R
Acta neuropathologica
126
329-52
2013
Show Abstract
β-Site amyloid precursor protein (APP) cleaving enzyme-1 (BACE1) is the β-secretase that initiates Aβ production in Alzheimer's disease (AD). BACE1 levels are increased in AD, which could contribute to pathogenesis, yet the mechanism of BACE1 elevation is unclear. Furthermore, the normal function of BACE1 is poorly understood. We localized BACE1 in the brain at both the light and electron microscopic levels to gain insight into normal and pathophysiologic roles of BACE1 in health and AD, respectively. Our findings provide the first ultrastructural evidence that BACE1 localizes to vesicles (likely endosomes) in normal hippocampal mossy fiber terminals of both non-transgenic and APP transgenic (5XFAD) mouse brains. In some instances, BACE1-positive vesicles were located near active zones, implying a function for BACE1 at the synapse. In addition, BACE1 accumulated in swollen dystrophic autophagosome-poor presynaptic terminals surrounding amyloid plaques in 5XFAD cortex and hippocampus. Importantly, accumulations of BACE1 and APP co-localized in presynaptic dystrophies, implying increased BACE1 processing of APP in peri-plaque regions. In primary cortical neuron cultures, treatment with the lysosomal protease inhibitor leupeptin caused BACE1 levels to increase; however, exposure of neurons to the autophagy inducer trehalose did not reduce BACE1 levels. This suggests that BACE1 is degraded by lysosomes but not by autophagy. Our results imply that BACE1 elevation in AD could be linked to decreased lysosomal degradation of BACE1 within dystrophic presynaptic terminals. Elevated BACE1 and APP levels in plaque-associated presynaptic dystrophies could increase local peri-plaque Aβ generation and accelerate amyloid plaque growth in AD. | | | 23820808
|
GABA(B) receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses.
Fiorentino, H; Kuczewski, N; Diabira, D; Ferrand, N; Pangalos, MN; Porcher, C; Gaiarsa, JL
The Journal of neuroscience : the official journal of the Society for Neuroscience
29
11650-61
2009
Show Abstract
GABA, the main inhibitory neurotransmitter in the adult brain, has recently emerged as an important signal in network development. Most of the trophic functions of GABA have been attributed to depolarization of the embryonic and neonatal neurons via the activation of ionotropic GABA(A) receptors. Here we demonstrate a novel mechanism by which endogenous GABA selectively regulates the development of GABAergic synapses in the developing brain. Using whole-cell patch-clamp recordings on newborn mouse hippocampi lacking functional GABA(B) receptors (GABA(B)-Rs) and time-lapse fluorescence imaging on cultured hippocampal neurons expressing GFP-tagged brain-derived neurotrophic factor (BDNF), we found that activation of metabotropic GABA(B) receptors (GABA(B)-Rs) triggers secretion of BDNF and promotes the development of perisomatic GABAergic synapses in the newborn mouse hippocampus. Because activation of GABA(B)-Rs occurs during the characteristic ongoing physiological network-driven synaptic activity present in the developing hippocampus, our results reveal a new mechanism by which synaptic activity can modulate the development of local GABAergic synaptic connections in the developing brain. | | | 19759312
|