Shh Signaling through the Primary Cilium Modulates Rat Oligodendrocyte Differentiation.
Falcón-Urrutia, P; Carrasco, CM; Lois, P; Palma, V; Roth, AD
PloS one
10
e0133567
2015
Show Abstract
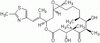
Primary Cilia (PC) are a very likely place for signal integration where multiple signaling pathways converge. Two major signaling pathways clearly shown to signal through the PC, Sonic Hedgehog (Shh) and PDGF-Rα, are particularly important for the proliferation and differentiation of oligodendrocytes, suggesting that their interaction occurs in or around this organelle. We identified PC in rat oligodendrocyte precursor cells (OPCs) and found that, while easily detectable in early OPCs, PC are lost as these cells progress to terminal differentiation. We confirmed the interaction between these pathways, as cyclopamine inhibition of Hedgehog function impairs both PDGF-mediated OPC proliferation and Shh-dependent cell branching. However, we failed to detect PDGF-Rα localization into the PC. Remarkably, ciliobrevin-mediated disruption of PC and reduction of OPC process extension was counteracted by recombinant Shh treatment, while PDGF had no effect. Therefore, while PDGF-Rα-dependent OPC proliferation and survival most probably does not initiate at the PC, still the integrity of this organelle and cilium-centered pathway is necessary for OPC survival and differentiation. | | | 26218245
 |
AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation.
Herms, A; Bosch, M; Reddy, BJ; Schieber, NL; Fajardo, A; Rupérez, C; Fernández-Vidal, A; Ferguson, C; Rentero, C; Tebar, F; Enrich, C; Parton, RG; Gross, SP; Pol, A
Nature communications
6
7176
2015
Show Abstract
Lipid droplets (LDs) are intracellular organelles that provide fatty acids (FAs) to cellular processes including synthesis of membranes and production of metabolic energy. While known to move bidirectionally along microtubules (MTs), the role of LD motion and whether it facilitates interaction with other organelles are unclear. Here we show that during nutrient starvation, LDs and mitochondria relocate on detyrosinated MT from the cell centre to adopt a dispersed distribution. In the cell periphery, LD-mitochondria interactions increase and LDs efficiently supply FAs for mitochondrial beta-oxidation. This cellular adaptation requires the activation of the energy sensor AMPK, which in response to starvation simultaneously increases LD motion, reorganizes the network of detyrosinated MTs and activates mitochondria. In conclusion, we describe the existence of a specialized cellular network connecting the cellular energetic status and MT dynamics to coordinate the functioning of LDs and mitochondria during nutrient scarcity. | Immunofluorescence | | 26013497
|
Using the avian mutant talpid2 as a disease model for understanding the oral-facial phenotypes of oral-facial-digital syndrome.
Schock, EN; Chang, CF; Struve, JN; Chang, YT; Chang, J; Delany, ME; Brugmann, SA
Disease models & mechanisms
8
855-66
2015
Show Abstract
Oral-facial-digital syndrome (OFD) is a ciliopathy that is characterized by oral-facial abnormalities, including cleft lip and/or palate, broad nasal root, dental anomalies, micrognathia and glossal defects. In addition, these individuals have several other characteristic abnormalities that are typical of a ciliopathy, including polysyndactyly, polycystic kidneys and hypoplasia of the cerebellum. Recently, a subset of OFD cases in humans has been linked to mutations in the centriolar protein C2 Ca(2+)-dependent domain-containing 3 (C2CD3). Our previous work identified mutations in C2CD3 as the causal genetic lesion for the avian talpid(2) mutant. Based on this common genetic etiology, we re-examined the talpid(2) mutant biochemically and phenotypically for characteristics of OFD. We found that, as in OFD-affected individuals, protein-protein interactions between C2CD3 and oral-facial-digital syndrome 1 protein (OFD1) are reduced in talpid(2) cells. Furthermore, we found that all common phenotypes were conserved between OFD-affected individuals and avian talpid(2) mutants. In light of these findings, we utilized the talpid(2) model to examine the cellular basis for the oral-facial phenotypes present in OFD. Specifically, we examined the development and differentiation of cranial neural crest cells (CNCCs) when C2CD3-dependent ciliogenesis was impaired. Our studies suggest that although disruptions of C2CD3-dependent ciliogenesis do not affect CNCC specification or proliferation, CNCC migration and differentiation are disrupted. Loss of C2CD3-dependent ciliogenesis affects the dispersion and directional persistence of migratory CNCCs. Furthermore, loss of C2CD3-dependent ciliogenesis results in dysmorphic and enlarged CNCC-derived facial cartilages. Thus, these findings suggest that aberrant CNCC migration and differentiation could contribute to the pathology of oral-facial defects in OFD. | Immunoblotting (Western) | | 26044959
|
Effects of α-tubulin K40 acetylation and detyrosination on kinesin-1 motility in a purified system.
Kaul, N; Soppina, V; Verhey, KJ
Biophysical journal
106
2636-43
2014
Show Abstract
Long-range transport in cells is achieved primarily through motor-based transport along a network of microtubule tracks. Targeted transport by kinesin motors can be correlated with posttranslational modifications (PTMs) of the tubulin subunits in specific microtubules. To directly examine the influence of specific PTMs on kinesin-1 motility, we generated tubulin subunits that were either enriched in or lacking acetylation of α-tubulin lysine 40 (K40) or detyrosination of the α-tubulin C-terminal tail. We show that K40 acetylation does not result in significant changes in kinesin-1's landing rate or motility parameters (velocity and run length) across experimental conditions. In contrast, detyrosination causes a moderate increase in kinesin-1's landing rate. The fact that the effects of detyrosination are dampened by prior K40 acetylation indicates that the combination of PTMs may be an important aspect of the functional output of microtubule heterogeneity. Importantly, our results indicate that the moderate influences that single PTMs have on kinesin-1 in vitro do not explain the strong correlation between specific PTMs and kinesin-1 transport in cells. Thus, additional mechanisms for regulating kinesin-1 transport in cells must be explored in future work. | | | 24940781
|
The cellular and molecular etiology of the craniofacial defects in the avian ciliopathic mutant talpid2.
Chang, CF; Schock, EN; O'Hare, EA; Dodgson, J; Cheng, HH; Muir, WM; Edelmann, RE; Delany, ME; Brugmann, SA
Development (Cambridge, England)
141
3003-12
2014
Show Abstract
talpid(2) is an avian autosomal recessive mutant with a myriad of congenital malformations, including polydactyly and facial clefting. Although phenotypically similar to talpid(3), talpid(2) has a distinct facial phenotype and an unknown cellular, molecular and genetic basis. We set out to determine the etiology of the craniofacial phenotype of this mutant. We confirmed that primary cilia were disrupted in talpid(2) mutants. Molecularly, we found disruptions in Hedgehog signaling. Post-translational processing of GLI2 and GLI3 was aberrant in the developing facial prominences. Although both GLI2 and GLI3 processing were disrupted in talpid(2) mutants, only GLI3 activator levels were significantly altered in the nucleus. Through additional fine mapping and whole-genome sequencing, we determined that the talpid(2) phenotype was linked to a 1.4 Mb region on GGA1q that contained the gene encoding the ciliary protein C2CD3. We cloned the avian ortholog of C2CD3 and found its expression was ubiquitous, but most robust in the developing limbs and facial prominences. Furthermore, we found that C2CD3 is localized proximal to the ciliary axoneme and is important for docking the mother centriole to the ciliary vesicle and cell membrane. Finally, we identified a 19 bp deletion in talpid(2) C2CD3 that produces a premature stop codon, and thus a truncated protein, as the likely causal allele for the phenotype. Together, these data provide insight into the cellular, molecular and genetic etiology of the talpid(2) phenotype. Our data suggest that, although the talpid(2) and talpid(3) mutations affect a common ciliogenesis pathway, they are caused by mutations in different ciliary proteins that result in differences in craniofacial phenotype. | Immunohistochemistry | | 25053433
|
ATMIN is a transcriptional regulator of both lung morphogenesis and ciliogenesis.
Goggolidou, P; Stevens, JL; Agueci, F; Keynton, J; Wheway, G; Grimes, DT; Patel, SH; Hilton, H; Morthorst, SK; DiPaolo, A; Williams, DJ; Sanderson, J; Khoronenkova, SV; Powles-Glover, N; Ermakov, A; Esapa, CT; Romero, R; Dianov, GL; Briscoe, J; Johnson, CA; Pedersen, LB; Norris, DP
Development (Cambridge, England)
141
3966-77
2014
Show Abstract
Initially identified in DNA damage repair, ATM-interactor (ATMIN) further functions as a transcriptional regulator of lung morphogenesis. Here we analyse three mouse mutants, Atmin(gpg6/gpg6), Atmin(H210Q/H210Q) and Dynll1(GT/GT), revealing how ATMIN and its transcriptional target dynein light chain LC8-type 1 (DYNLL1) are required for normal lung morphogenesis and ciliogenesis. Expression screening of ciliogenic genes confirmed Dynll1 to be controlled by ATMIN and further revealed moderately altered expression of known intraflagellar transport (IFT) protein-encoding loci in Atmin mutant embryos. Significantly, Dynll1(GT/GT) embryonic cilia exhibited shortening and bulging, highly similar to the characterised retrograde IFT phenotype of Dync2h1. Depletion of ATMIN or DYNLL1 in cultured cells recapitulated the in vivo ciliogenesis phenotypes and expression of DYNLL1 or the related DYNLL2 rescued the effects of loss of ATMIN, demonstrating that ATMIN primarily promotes ciliogenesis by regulating Dynll1 expression. Furthermore, DYNLL1 as well as DYNLL2 localised to cilia in puncta, consistent with IFT particles, and physically interacted with WDR34, a mammalian homologue of the Chlamydomonas cytoplasmic dynein 2 intermediate chain that also localised to the cilium. This study extends the established Atmin-Dynll1 relationship into a developmental and a ciliary context, uncovering a novel series of interactions between DYNLL1, WDR34 and ATMIN. This identifies potential novel components of cytoplasmic dynein 2 and furthermore provides fresh insights into the molecular pathogenesis of human skeletal ciliopathies. | | | 25294941
|
A Mec17-Myosin II Effector Axis Coordinates Microtubule Acetylation and Actin Dynamics to Control Primary Cilium Biogenesis.
Rao, Y; Hao, R; Wang, B; Yao, TP
PloS one
9
e114087
2014
Show Abstract
Primary cilia are specialized, acetylated microtubule-based signaling processes. Cilium assembly is activated by cellular quiescence and requires reconfiguration of microtubules, the actin cytoskeleton, and vesicular trafficking machinery. How these components are coordinated to activate ciliogenesis remains unknown. Here we identify the microtubule acetyltransferase Mec-17 and myosin II motors as the key effectors in primary cilium biogenesis. We found that myosin IIB (Myh10) is required for cilium formation; however, myosin IIA (Myh9) suppresses it. Myh10 binds and antagonizes Myh9 to increase actin dynamics, which facilitates the assembly of the pericentrosomal preciliary complex (PPC) that supplies materials for cilium growth. Importantly, Myh10 expression is upregulated by serum-starvation and this induction requires Mec-17, which is itself accumulated upon cellular quiescence. Pharmacological stimulation of microtubule acetylation also induces Myh10 expression and cilium formation. Thus cellular quiescence induces Mec17 to couple the production of acetylated microtubules and Myh10, whose accumulation overcomes the inhibitory role of Myh9 and initiates ciliogenesis. | | | 25494100
|
Kif3a guides microtubular dynamics, migration and lumen formation of MDCK cells.
Boehlke, C; Kotsis, F; Buchholz, B; Powelske, C; Eckardt, KU; Walz, G; Nitschke, R; Kuehn, EW
PloS one
8
e62165
2013
Show Abstract
The microtubular motor Kinesin-2 and its subunit Kif3a are essential for the formation of primary cilia, an organelle implicated in a wide spectrum of developmental abnormalities. Outside cilia, Kinesin-2 mediated transport has been implicated in vesicle and N-cadherin transport, but it is unknown if and how extraciliary Kif3a affects basic cellular functions such as migration or the formation of multicellular structures. Here we show that tetracycline inducible depletion of Kif3a in MDCK cells slows epithelial cell migration. Microtubules at the leading edge of Kif3a depleted cells failed to grow perpendicularly into the leading edge and microtubular dynamics were dampened in Kif3a depleted cells. Loss of Kif3a retarded lateral membrane specification and completely prevented the formation of three-dimensional spheres in collagen. These data uncover that Kif3a regulates the microtubular cytoskeleton in the cell periphery and imply that extra-ciliary Kif3a has an unexpected function in morphogenesis. | Western Blotting | | 23658710
|
α2-Chimaerin regulates a key axon guidance transition during development of the oculomotor projection.
Clark, C; Austen, O; Poparic, I; Guthrie, S
The Journal of neuroscience : the official journal of the Society for Neuroscience
33
16540-51
2013
Show Abstract
The ocular motor system consists of three nerves which innervate six muscles to control eye movements. In humans, defective development of this system leads to eye movement disorders, such as Duane Retraction Syndrome, which can result from mutations in the α2-chimaerin signaling molecule. We have used the zebrafish to model the role of α2-chimaerin during development of the ocular motor system. We first mapped ocular motor spatiotemporal development, which occurs between 24 and 72 h postfertilization (hpf), with the oculomotor nerve following an invariant sequence of growth and branching to its muscle targets. We identified 52 hpf as a key axon guidance "transition," when oculomotor axons reach the orbit and select their muscle targets. Live imaging and quantitation showed that, at 52 hpf, axons undergo a switch in behavior, with striking changes in the dynamics of filopodia. We tested the role of α2-chimaerin in this guidance process and found that axons expressing gain-of-function α2-chimaerin isoforms failed to undergo the 52 hpf transition in filopodial dynamics, leading to axon stalling. α2-chimaerin loss of function led to ecotopic and misguided branching and hypoplasia of oculomotor axons; embryos had defective eye movements as measured by the optokinetic reflex. Manipulation of chimaerin signaling in oculomotor neurons in vitro led to changes in microtubule stability. These findings demonstrate that a correct level of α2-chimaerin signaling is required for key oculomotor axon guidance decisions, and provide a zebrafish model for Duane Retraction Syndrome. | | | 24133258
|
aPKCλ/ι and aPKCζ contribute to podocyte differentiation and glomerular maturation.
Hartleben, B; Widmeier, E; Suhm, M; Worthmann, K; Schell, C; Helmstädter, M; Wiech, T; Walz, G; Leitges, M; Schiffer, M; Huber, TB
Journal of the American Society of Nephrology : JASN
24
253-67
2013
Show Abstract
Precise positioning of the highly complex interdigitating podocyte foot processes is critical to form the normal glomerular filtration barrier, but the molecular programs driving this process are unknown. The protein atypical protein kinase C (aPKC)--a component of the Par complex, which localizes to tight junctions and interacts with slit diaphragm proteins--may play a role. Here, we found that the combined deletion of the aPKCλ/ι and aPKCζ isoforms in podocytes associated with incorrectly positioned centrosomes and Golgi apparatus and mislocalized molecules of the slit diaphragm. Furthermore, aPKC-deficient podocytes failed to form the normal network of foot processes, leading to defective glomerular maturation with incomplete capillary formation and mesangiolysis. Our results suggest that aPKC isoforms orchestrate the formation of the podocyte processes essential for normal glomerular development and kidney function. Defective aPKC signaling results in a dramatically simplified glomerular architecture, causing severe proteinuria and perinatal death. | | | 23334392
|