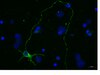
An autism-associated variant of Epac2 reveals a role for Ras/Epac2 signaling in controlling basal dendrite maintenance in mice.
Srivastava, Deepak P, et al.
PLoS Biol., 10: e1001350 (2012)
2011
Abstract anzeigen
The architecture of dendritic arbors determines circuit connectivity, receptive fields, and computational properties of neurons, and dendritic structure is impaired in several psychiatric disorders. While apical and basal dendritic compartments of pyramidal neurons are functionally specialized and differentially regulated, little is known about mechanisms that selectively maintain basal dendrites. Here we identified a role for the Ras/Epac2 pathway in maintaining basal dendrite complexity of cortical neurons. Epac2 is a guanine nucleotide exchange factor (GEF) for the Ras-like small GTPase Rap, and it is highly enriched in the adult mouse brain. We found that in vivo Epac2 knockdown in layer 2/3 cortical neurons via in utero electroporation reduced basal dendritic architecture, and that Epac2 knockdown in mature cortical neurons in vitro mimicked this effect. Overexpression of an Epac2 rare coding variant, found in human subjects diagnosed with autism, also impaired basal dendritic morphology. This mutation disrupted Epac2's interaction with Ras, and inhibition of Ras selectively interfered with basal dendrite maintenance. Finally, we observed that components of the Ras/Epac2/Rap pathway exhibited differential abundance in the basal versus apical dendritic compartments. These findings define a role for Epac2 in enabling crosstalk between Ras and Rap signaling in maintaining basal dendrite complexity, and exemplify how rare coding variants, in addition to their disease relevance, can provide insight into cellular mechanisms relevant for brain connectivity. | 22745599
 |
Social, Communication, and Cortical Structural Impairments in Epac2-Deficient Mice.
Srivastava, Deepak P, et al.
J. Neurosci., 32: 11864-11878 (2012)
2011
Abstract anzeigen
Deficits in social and communication behaviors are common features of a number of neurodevelopmental disorders. However, the molecular and cellular substrates of these higher order brain functions are not well understood. Here we report that specific alterations in social and communication behaviors in mice occur as a result of loss of the EPAC2 gene, which encodes a protein kinase A-independent cAMP target. Epac2-deficient mice exhibited robust deficits in social interactions and ultrasonic vocalizations, but displayed normal olfaction, working and reference memory, motor abilities, anxiety, and repetitive behaviors. Epac2-deficient mice displayed abnormal columnar organization in the anterior cingulate cortex, a region implicated in social behavior in humans, but not in somatosensory cortex. In vivo two-photon imaging revealed reduced dendritic spine motility and density on cortical neurons in Epac2-deficient mice, indicating deficits at the synaptic level. Together, these findings provide novel insight into the molecular and cellular substrates of social and communication behavior. | 22915127
|
PKA and Epac synergistically inhibit smooth muscle cell proliferation.
Hewer, RC; Sala-Newby, GB; Wu, YJ; Newby, AC; Bond, M
Journal of molecular and cellular cardiology
50
87-98
2010
Abstract anzeigen
Cyclic AMP signalling promotes VSMC quiescence in healthy vessels and during vascular healing following injury. Cyclic AMP inhibits VSMC proliferation via mechanisms that are not fully understood. We investigated the role of PKA and Epac signalling on cAMP-induced inhibition of VSMC proliferation. cAMP-mediated growth arrest was PKA-dependent. However, selective PKA activation with 6-Benzoyl-cAMP did not inhibit VSMC proliferation, indicating a requirement for additional pathways. Epac activation using the selective cAMP analogue 8-CPT-2'-O-Me-cAMP, did not affect levels of hyperphosphorylated Retinoblastoma (Rb) protein, a marker of G1-S phase transition, or BrdU incorporation, despite activation of the Epac-effector Rap1. However, 6-Benzoyl-cAMP and 8-CPT-2'-O-Me-cAMP acted synergistically to inhibit Rb-hyperphosphorylation and BrdU incorporation, indicating that both pathways are required for growth inhibition. Consistent with this, constitutively active Epac increased Rap1 activity and synergised with 6-Benzoyl-cAMP to inhibit VSMC proliferation. PKA and Epac synergised to inhibit phosphorylation of ERK and JNK. Induction of stellate morphology, previously associated with cAMP-mediated growth arrest, was also dependent on activation of both PKA and Epac. Rap1 inhibition with Rap1GAP or siRNA silencing did not negate forskolin-induced inhibition of Rb-hyperphosphorylation, BrdU incorporation or stellate morphology. This data demonstrates for the first time that Epac synergises with PKA via a Rap1-independent mechanism to mediate cAMP-induced growth arrest in VSMC. This work highlights the role of Epac as a major player in cAMP-dependent growth arrest in VSMC. | 20971121
|
Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers.
Cheung, HW; Du, J; Boehm, JS; He, F; Weir, BA; Wang, X; Butaney, M; Sequist, LV; Luo, B; Engelman, JA; Root, DE; Meyerson, M; Golub, TR; Jänne, PA; Hahn, WC
Cancer discovery
1
608-25
2010
Abstract anzeigen
We previously identified a region of recurrent amplification on chromosome 22q11.21 in a subset of primary lung adenocarcinomas. Here we show that CRKL, encoding for an adaptor protein, is amplified and overexpressed in non-small cell lung cancer (NSCLC) cells that harbor 22q11.21 amplifications. Overexpression of CRKL in immortalized human airway epithelial cells promoted anchorage-independent growth and tumorigenicity. Oncogenic CRKL activates the SOS1-RAS-RAF-ERK and SRC-C3G-RAP1 pathways. Suppression of CRKL in NSCLC cells that harbor CRKL amplifications induced cell death. Overexpression of CRKL in epidermal growth factor receptor (EGFR)-mutant cells induces resistance to gefitinib by activating extracellular signal-regulated kinase and AKT signaling. We identified CRKL amplification in an EGFR inhibitor-treated lung adenocarcinoma that was not present before treatment. These observations demonstrate that CRKL overexpression induces cell transformation, credential CRKL as a therapeutic target for a subset of NSCLC that harbor CRKL amplifications, and implicate CRKL as an additional mechanism of resistance to EGFR-directed therapy.These studies credential CRKL as an oncogene in a subset of NSCLC. Overexpression of CRKL induces cell transformation and resistance to epidermal growth factor receptor inhibitor treatment and suggest that therapeutic interventions targeting CRKL may confer a clinical benefit in a defined subset of NSCLCs. | 22586683
|
Possible role of the exchange protein directly activated by cyclic AMP (Epac) in the cyclic AMP-dependent functional differentiation and syncytialization of human placental BeWo cells.
Yoshie, M; Kaneyama, K; Kusama, K; Higuma, C; Nishi, H; Isaka, K; Tamura, K
Human reproduction (Oxford, England)
25
2229-38
2009
Abstract anzeigen
The mononuclear villous cytotrophoblast (CTB) differentiates and fuses to the multinucleated syncytiotrophoblast (STB), which produces hCG and progesterone. cAMP-mediated intracellular pathways are involved in the process of endocrine differentiation and fusion (syncytialization). The exchange protein directly activated by cAMP (Epac) is a mediator of cAMP signaling. We examined the differential roles of Epac and protein kinase A (PKA) signaling in the cell fusion and differentiation of trophoblast-derived BeWo cells.Epac1 and Epac2 were localized in human placental tissue (n = 9) by immunohistochemistry. The PKA-selective cAMP analog (N(6)-phenyl-cAMP, Phe) or Epac-selective cAMP analog (CPT) was tested for effects on hCG and progesterone production, and syncytialization in BeWo cells. The effect of knockdown of Epac or its downstream target molecule (Rap1) on syncytialization was evaluated.Epac1 and Epac2 proteins were expressed in villous CTB, STB, stroma, blood vessels and extravillous CTB of the placenta. Phe increased the expression of hCG alpha/beta mRNA and secretion of hCG protein in BeWo cells (P less than 0.01 versus control). CPT-stimulated production of hCG (P less than 0.05), albeit to a lesser extent than Phe. Progesterone production was also enhanced by Phe or CPT (P less than 0.01 and P less than 0.05, respectively). CPT or a stable cAMP analog (dibutyryl-cAMP: Db) increased the number of syncytialized BeWo cells (P less than 0.01), whereas Phe did not stimulate fusion. CPT- or Db-induced syncytialization was observed, even in the presence of a PKA inhibitor. Knockdown of Epac1 or Rap1 repressed the Db-, CPT- or forskolin-induced cell fusion.The Epac signaling pathway may be associated with the cAMP-mediated functional differentiation and syncytialization of human trophoblasts. | 20663796
|
Driven to death: Inhibition of farnesylation increases Ras activity and promotes growth arrest and cell death [corrected].
Geryk-Hall, M; Yang, Y; Hughes, DP
Molecular cancer therapeutics
9
1111-9
2009
Abstract anzeigen
To improve cancer outcomes, investigators are turning increasingly to small molecule medicines that disrupt vital signaling cascades, inhibit malignant growth, or induce apoptosis. One vital signaling molecule is Ras, and a key step in Ras activation is membrane anchoring of Ras through prenylation, the C-terminal addition of a lipid anchor. Small molecule inhibitors of farnesyltransferase (FTI), the enzyme most often responsible for prenylating Ras, showed clinical promise, but development of FTIs such as tipifarnib has been stalled by uncertainty about their mechanism of action, because Ras seemed unimpeded in tipifarnib-treated samples. Interpretation was further complicated by the numerous proteins that may be farnesylated, as well as availability of an alternate prenylation pathway, geranylgeranylation. Our initial observations of varied response by cancer cell lines to tipifarnib led us to evaluate the role of FTI in Ras signal alteration using various tumor models. We describe our novel counterintuitive finding that endogenous Ras activity increases in cancer cell lines with low endogenous Ras activity when farnesyltransferase is inhibited by either tipifarnib or short hairpin RNA. In response to tipifarnib, variable growth arrest and/or cell death correlated with levels of activated extracellular signal–regulated kinase (ERK) and p38 mitogenactivated protein kinase (MAPK). Sensitivity to tipifarnib treatment was shown by growth inhibition and by an increase in subdiploid cell numbers; cells with such sensitivity had increased activation of ERK and p38 MAPK. Because Ras must be prenylated to be active, our findings suggest that geranylgeranylated N-Ras or K-Ras B interacts differently with downstream effector proteins in sensitive cancer cells responding to tipifarnib, switching the balance from cell proliferation to growth inhibition [corrected]. | 20406948
|