Wenn Sie das Fenster schließen, wird Ihre Konfiguration nicht gespeichert, es sei denn, Sie haben Ihren Artikel in die Bestellung aufgenommen oder zu Ihren Favoriten hinzugefügt.
Klicken Sie auf OK, um das MILLIPLEX® MAP-Tool zu schließen oder auf Abbrechen, um zu Ihrer Auswahl zurückzukehren.
Wählen Sie konfigurierbare Panels & Premixed-Kits - ODER - Kits für die zelluläre Signaltransduktion & MAPmates™
Konfigurieren Sie Ihre MILLIPLEX® MAP-Kits und lassen sich den Preis anzeigen.
Konfigurierbare Panels & Premixed-Kits
Unser breites Angebot enthält Multiplex-Panels, für die Sie die Analyten auswählen können, die am besten für Ihre Anwendung geeignet sind. Unter einem separaten Register können Sie das Premixed-Cytokin-Format oder ein Singleplex-Kit wählen.
Kits für die zelluläre Signaltransduktion & MAPmates™
Wählen Sie gebrauchsfertige Kits zur Erforschung gesamter Signalwege oder Prozesse. Oder konfigurieren Sie Ihre eigenen Kits mit Singleplex MAPmates™.
Die folgenden MAPmates™ sollten nicht zusammen analysiert werden: -MAPmates™, die einen unterschiedlichen Assaypuffer erfordern. -Phosphospezifische und MAPmate™ Gesamtkombinationen wie Gesamt-GSK3β und Gesamt-GSK3β (Ser 9). -PanTyr und locusspezifische MAPmates™, z.B. Phospho-EGF-Rezeptor und Phospho-STAT1 (Tyr701). -Mehr als 1 Phospho-MAPmate™ für ein einziges Target (Akt, STAT3). -GAPDH und β-Tubulin können nicht mit Kits oder MAPmates™, die panTyr enthalten, analysiert werden.
.
Bestellnummer
Bestellinformationen
St./Pkg.
Liste
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Wählen Sie bitte Spezies, Panelart, Kit oder Probenart
Um Ihr MILLIPLEX® MAP-Kit zu konfigurieren, wählen Sie zunächst eine Spezies, eine Panelart und/oder ein Kit.
Custom Premix Selecting "Custom Premix" option means that all of the beads you have chosen will be premixed in manufacturing before the kit is sent to you.
Catalogue Number
Ordering Description
Qty/Pack
List
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Spezies
Panelart
Gewähltes Kit
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
96-Well Plate
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
Weitere Reagenzien hinzufügen (MAPmates erfordern die Verwendung eines Puffer- und Detektionskits)
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
48-602MAG
Buffer Detection Kit for Magnetic Beads
1 Kit
Platzsparende Option Kunden, die mehrere Kits kaufen, können ihre Multiplex-Assaykomponenten in Kunststoffbeuteln anstelle von Packungen erhalten, um eine kompaktere Lagerung zu ermöglichen.
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Das Produkt wurde in Ihre Bestellung aufgenommen
Sie können nun ein weiteres Kit konfigurieren, ein Premixed-Kit wählen, zur Kasse gehen oder das Bestell-Tool schließen.
AB5058
Sigma-AldrichAnti-Prion Protein Antibody, NT, a.a. 78-97
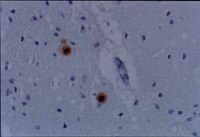
Anti-Prion Protein Antibody, N-terminus, a.a. 78-97 detects level of Prion Protein & has been published & validated for use in ELISA, WB, IH(P).
More>>Anti-Prion Protein Antibody, N-terminus, a.a. 78-97 detects level of Prion Protein & has been published & validated for use in ELISA, WB, IH(P). Less<<
Anti-Prion Protein Antibody, NT, a.a. 78-97: SDB (Sicherheitsdatenblätter), Analysenzertifikate und Qualitätszertifikate, Dossiers, Broschüren und andere verfügbare Dokumente.
Anti-Prion Protein Antibody, N-terminus, a.a. 78-97 detects level of Prion Protein & has been published & validated for use in ELISA, WB, IH(P).
Key Applications
ELISA
Western Blotting
Immunohistochemistry (Paraffin)
Application Notes
Immunohistochemistry: >1:200 on paraffin embedded, formalin fixed human brain.
Western blots: > 1:2,000
ELISA: > 1:35,000
Optimal working dilutions must be determined by the end user.
Biological Information
Immunogen
Synthetic peptide that corresponds to amino acids 79-97 of the N-terminus of the human PrP27-30.
Epitope
N-terminus, a.a. 78-97
Host
Goat
Specificity
Specific for human prion protein (PrP). This antibody immunolabels amyloid plaques in formalin-fixed paraffin sections from Creutzfeld-Jakob Disease (CJD) brain.The prion protein is a large membrane protein that occurs normally in neurons of the human brain and is thought to be involved in synaptic transmission. In prion diseases, such as CJD, Gerstmann-Straussler-Scheinker syndrome (GSS), Fatal Familial Insomnia (FFI), Alpers Syndrome and Kuru, the normal cellular form of this protein (PrPc) is transformed into an altered protein when it comes into contact with an infectious prion protein (PrPsc) from another host. This altered PrPsc accumulates in cytoplasmic vesicles of diseased individuals forming lesions, vacuoles and amyloid deposits.
The protein encoded by this gene is a membrane glycosylphosphatidylinositol-anchored glycoprotein that tends to aggregate into rod-like structures. The encoded protein contains a highly unstable region of five tandem octapeptide repeats. This gene is found on chromosome 20, approximately 20 kbp upstream of a gene which encodes a biochemically and structurally similar protein to the one encoded by this gene. Mutations in the repeat region as well as elsewhere in this gene have been associated with Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Straussler disease, Huntington disease-like 1, and kuru. Alternative splicing results in multiple transcript variants encoding the same protein.
FUNCTION: SwissProt: P04156 # The physiological function of PrP is not known. SIZE: 253 amino acids; 27661 Da SUBUNIT: PrP has a tendency to aggregate yielding polymers called rods. SUBCELLULAR LOCATION: Cell membrane; Lipid-anchor, GPI-anchor. PTM: The glycosylation pattern (the amount of mono-, di- and non- glycosylated forms or glycoforms) seems to differ in normal and CJD prion. DISEASE: SwissProt: P04156 # PrP is found in high quantity in the brain of humans and animals infected with neurodegenerative diseases known as transmissible spongiform encephalopathies or prion diseases, like: Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), Gerstmann-Straussler disease (GSD), Huntington disease-like 1 (HDL1) and kuru in humans; scrapie in sheep and goat; bovine spongiform encephalopathy (BSE) in cattle; transmissible mink encephalopathy (TME); chronic wasting disease (CWD) of mule deer and elk; feline spongiform encephalopathy (FSE) in cats and exotic ungulate encephalopathy (EUE) in nyala and greater kudu. The prion diseases illustrate three manifestations of CNS degeneration: (1) infectious (2) sporadic and (3) dominantly inherited forms. TME, CWD, BSE, FSE, EUE are all thought to occur after consumption of prion-infected foodstuffs. & Defects in PRNP are the cause of Creutzfeldt-Jakob disease (CJD) [MIM:123400]. CJD occurs primarily as a sporadic disorder (1 per million), while 10-15% are familial. Accidental transmission of CJD to humans appears to be iatrogenic (contaminated human growth hormone (HGH), corneal transplantation, electroencephalographic electrode implantation, etc.). Epidemiologic studies have failed to implicate the ingestion of infected annimal meat in the pathogenesis of CJD in human. The triad of microscopic features that characterize the prion diseases consists of (1) spongiform degeneration of neurons, (2) severe astrocytic gliosis that often appears to be out of proportion to the degree of nerve cell loss, and (3) amyloid plaque formation. CJD is characterized by progressive dementia and myoclonic seizures, affecting adults in mid-life. Some patients present sleep disorders, abnormalities of high cortical function, cerebellar and corticospinal disturbances. The disease ends in death after a 3-12 months illness. & Defects in PRNP are the cause of fatal familial insomnia (FFI) [MIM:600072]. FFI is an autosomal dominant disorder and is characterized by neuronal degeneration limited to selected thalamic nuclei and progressive insomnia. & Defects in PRNP are the cause of Gerstmann-Straussler disease (GSD) [MIM:137440]. GSD is a heterogeneous disorder and was defined as a spinocerebellar ataxia with dementia and plaquelike deposits. GSD incidence is less than 2 per 100 million live births. & Defects in PRNP are the cause of Huntington disease-like 1 (HDL1) [MIM:603218]. HDL1 is an autosomal dominant, early onset neurodegenerative disorder with prominent psychiatric features. & Defects in PRNP are the cause of kuru [MIM:245300]. Kuru is transmitted during ritualistic cannibalism, among natives of the New Guinea highlands. Patients exhibit various movement disorders like cerebellar abnormalities, rigidity of the limbs, and clonus. Emotional lability is present, and dementia is conspicuously absent. Death usually occurs from 3 to 12 month after onset. & Defects in PRNP are the cause of prion disease with protracted course [MIM:606688]; an autosomal dominant presenile dementia with a rapidly progressive and protracted clinical course. The dementia was characterized clinically by frontotemporal features, including early personality changes. Some patients had memory loss, several showed aggressiveness, hyperorality and verbal stereotypy, others had parkinsonian symptoms.SIMILARITY:SwissProt: P04156 ## Belongs to the prion family.
Physicochemical Information
Dimensions
Materials Information
Toxicological Information
Safety Information according to GHS
Safety Information
Product Usage Statements
Usage Statement
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
Storage and Shipping Information
Storage Conditions
Maintain at -20°C in undiluted aliquots for up to 12 months. Avoid repeated freeze/thaw cycles.
Creutzfeldt-Jakob disease in Mexico. Leora Velásquez-Pérez, Daniel Rembao-Bojorquez, Jorge Guevara, Rosa María Guadarrama-Torres, Araceli Trejo-Contreras Neuropathology : official journal of the Japanese Society of Neuropathology
27
419-28
2007
Creutzfeldt-Jakob disease (CJD) is classified within the group of transmissible spongiform encephalopathies (TSE). It is a rapidly progressive illness that affects mental functions. The average age of onset is 50 years. Various tests can help orient the clinical diagnosis, but the confirmatory test is still the post mortem analysis. The aim of this study was to describe the epidemiological, clinical and histopathological characteristics of patients diagnosed as suffering from CJD, at the National Institute of Neurology and Neurosurgery of Mexico (NINN). An observational, descriptive and transversal study was conducted. We collected information concerning these cases from the Departments of Epidemiology and Pathology, as well as the clinical charts of the patients with a diagnosis of CJD. Fifteen cases were registered of which three CJD cases were definite, five probable cases were identified, and seven were possible. The average age of the patients was 49 years. Two definite cases were female and one was male. It is important to improve the systems for surveillance of this type of disease and, furthermore, to permit greater accessibility to laboratories where the procedures necessary for supporting diagnosis can be followed.
Molecular and genetic features of a labeled class of spinal substantia gelatinosa neurons in a transgenic mouse. Adam W Hantman, Edward R Perl The Journal of comparative neurology
492
90-100
2004
Genetic incorporation in a mouse of a transgene containing the prion promoter and the green fluorescent protein (GFP) coding sequence labels a set of substantia gelatinosa (SG) neurons (SG-GFP) homogenous in morphology, electrophysiology, and gamma-amino-butyric acid expression. In the present analysis the SG-GFP neurons are established to have protein kinase C-betaII immunoreactivity and to lack evidence for the presence of calbindin D-28k, parvalbumin, and protein kinase C-gamma. These neurons were hyperpolarized by mediators of descending control, norepinephrine and serotonin. Sequential polymerase chain reactions established the insertion of the transgene to be in the receptor protein tyrosine phosphatase kappa (RPTP-kappa) and the laminin receptor 1 (ribosomal protein SA) pseudogene 1 locus. RPTP-kappa expression in both GFP-labeled dorsal root ganglia and SG neurons raises the possibility that homophilic interactions of RPTP-kappa contribute to establishment of connections between specific classes of primary afferent and SG neurons.
Toluidine blue-O staining of prion protein deposits. A Sánchez, A Guzmán, A Ortiz, D Rembao, B Espinosa, E Zenteno, J Guevara Histochemistry and cell biology
116
519-24
2001
Prion diseases or transmissible spongiform encephalopathies are a group of fatal neurodegenerative diseases caused by an abnormal form of prion protein (PrP(sc)). In this study, we developed a sensitive histochemical detection of PrP(sc) deposits in a Gertsmann-Sträussler-Scheinker disease (GSS) patient using toluidine blue-O staining, a specific reagent to stain mucins and mucopolysaccharides. Detection of prion deposits correlated with immunohistochemistry using anti-prion antibodies. Control assays were performed using amyloid-beta (Abeta) plaques from Alzheimer's disease (AD) brains. Our results demonstrated that toluidine blue-O staining allowed to recognize 69.1+/-2.6% of the total plaques recognized by the anti-prion antibody. Furthermore, in the 15 studied brain regions from the GSS patient, toluidine blue-O revealed the same recognition pattern as anti-prion labeling. Toluidine blue-O stained specifically the prion deposits but not the Abeta plaques in AD brains. The specificity of the technique was confirmed in a Creutzfeldt-Jakob disease brain. This method opens several possibilities for postmortem diagnoses. Our results also suggest the relevance of specific post-translational modifications of PrP(sc), identified by toluidine blue-O, that might participate in the transformation of PrP(c) to PrP(sc).
Copper has differential effect on prion protein with polymorphism of position 129. B S Wong, C Clive, S J Haswell, R A Williamson, D R Burton, P Gambetti, M S Sy, I M Jones, D R Brown Biochemical and biophysical research communications
269
726-31
1999
The pathology of human prion diseases is affected by polymorphism at amino acid residue 129 of the prion protein gene. Recombinant mouse prion proteins mimicking either form of the polymorphism were prepared to examine their effect on the conformation and the level of superoxide dismutase (SOD) activity of the prion protein. Following the binding of copper atoms to prion protein, antibody mapping and CD analysis detected conformational differences between the two forms of protein. However, neither the level of copper binding nor the level of SOD activity associated with this form of prion protein altered with the identity of codon 129. These results suggest that in the holo-metal binding form of the protein, prion structure but not its SOD activity is affected by polymorphism at codon 129.
Selective oxidation of methionine residues in prion proteins. B S Wong, H Wang, D R Brown, I M Jones Biochemical and biophysical research communications
259
352-5
1998
Prion proteins are central to the pathogenesis of several neurodegenerative diseases through the postulated conversion of the endogenous cellular isoform (PrPc) into a pathogenic isoform (PrPSc). Although the cellular function of normal prion protein remains unresolved a number of studies have shown that prion proteins may be involved in the cellular response to oxidative stress. Here, using purified recombinant sources of mouse and chicken PrP refolded in the presence of copper (II) we show that the methionine residues of the protein are uniquely susceptible to oxidation. We suggest that Met residues may form an essential part of the mechanism of the antioxidant activity exhibited by normal prion protein.