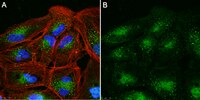
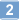Localization of complement factor H gene expression and protein distribution in the mouse outer retina.
Smit-McBride, Z; Oltjen, SL; Radu, RA; Estep, J; Nguyen, AT; Gong, Q; Hjelmeland, LM
Molecular vision
21
110-23
2015
요약 표시
To determine the localization of complement factor H (Cfh) mRNA and its protein in the mouse outer retina.Quantitative real-time PCR (qPCR) was used to determine the expression of Cfh and Cfh-related (Cfhr) transcripts in the RPE/choroid. In situ hybridization (ISH) was performed using the novel RNAscope 2.0 FFPE assay to localize the expression of Cfh mRNA in the mouse outer retina. Immunohistochemistry (IHC) was used to localize Cfh protein expression, and western blots were used to characterize CFH antibodies used for IHC.Cfh and Cfhr2 transcripts were detected in the mouse RPE/choroid using qPCR, while Cfhr1, Cfhr3, and Cfhrc (Gm4788) were not detected. ISH showed abundant Cfh mRNA in the RPE of all mouse strains (C57BL/6, BALB/c, 129/Sv) tested, with the exception of the Cfh(-/-) eye. Surprisingly, the Cfh protein was detected by immunohistochemistry in photoreceptors rather than in RPE cells. The specificity of the CFH antibodies was tested by western blotting. Our CFH antibodies recognized purified mouse Cfh protein, serum Cfh protein in wild-type C57BL/6, BALB/c, and 129/Sv, and showed an absence of the Cfh protein in the serum of Cfh(-/-) mice. Greatly reduced Cfh protein immunohistological signals in the Cfh(-/-) eyes also supported the specificity of the Cfh protein distribution results.Only Cfh and Cfhr2 genes are expressed in the mouse outer retina. Only Cfh mRNA was detected in the RPE, but no protein. We hypothesize that the steady-state concentration of Cfh protein is low in the cells due to secretion, and therefore is below the detection level for IHC. | | | 25684976
 |
CRALBP supports the mammalian retinal visual cycle and cone vision.
Xue, Y; Shen, SQ; Jui, J; Rupp, AC; Byrne, LC; Hattar, S; Flannery, JG; Corbo, JC; Kefalov, VJ
The Journal of clinical investigation
125
727-38
2015
요약 표시
Mutations in the cellular retinaldehyde-binding protein (CRALBP, encoded by RLBP1) can lead to severe cone photoreceptor-mediated vision loss in patients. It is not known how CRALBP supports cone function or how altered CRALBP leads to cone dysfunction. Here, we determined that deletion of Rlbp1 in mice impairs the retinal visual cycle. Mice lacking CRALBP exhibited M-opsin mislocalization, M-cone loss, and impaired cone-driven visual behavior and light responses. Additionally, M-cone dark adaptation was largely suppressed in CRALBP-deficient animals. While rearing CRALBP-deficient mice in the dark prevented the deterioration of cone function, it did not rescue cone dark adaptation. Adeno-associated virus-mediated restoration of CRALBP expression specifically in Müller cells, but not retinal pigment epithelial (RPE) cells, rescued the retinal visual cycle and M-cone sensitivity in knockout mice. Our results identify Müller cell CRALBP as a key component of the retinal visual cycle and demonstrate that this pathway is important for maintaining normal cone-driven vision and accelerating cone dark adaptation. | | | 25607845
|
Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia.
Kohl, S; Zobor, D; Chiang, WC; Weisschuh, N; Staller, J; Gonzalez Menendez, I; Chang, S; Beck, SC; Garcia Garrido, M; Sothilingam, V; Seeliger, MW; Stanzial, F; Benedicenti, F; Inzana, F; Héon, E; Vincent, A; Beis, J; Strom, TM; Rudolph, G; Roosing, S; Hollander, AI; Cremers, FP; Lopez, I; Ren, H; Moore, AT; Webster, AR; Michaelides, M; Koenekoop, RK; Zrenner, E; Kaufman, RJ; Tsang, SH; Wissinger, B; Lin, JH
Nature genetics
47
757-65
2015
요약 표시
Achromatopsia (ACHM) is an autosomal recessive disorder characterized by color blindness, photophobia, nystagmus and severely reduced visual acuity. Using homozygosity mapping and whole-exome and candidate gene sequencing, we identified ten families carrying six homozygous and two compound-heterozygous mutations in the ATF6 gene (encoding activating transcription factor 6A), a key regulator of the unfolded protein response (UPR) and cellular endoplasmic reticulum (ER) homeostasis. Patients had evidence of foveal hypoplasia and disruption of the cone photoreceptor layer. The ACHM-associated ATF6 mutations attenuate ATF6 transcriptional activity in response to ER stress. Atf6(-/-) mice have normal retinal morphology and function at a young age but develop rod and cone dysfunction with increasing age. This new ACHM-related gene suggests a crucial and unexpected role for ATF6A in human foveal development and cone function and adds to the list of genes that, despite ubiquitous expression, when mutated can result in an isolated retinal photoreceptor phenotype. | Western Blotting | | 26029869
|
AAV-mediated RLBP1 gene therapy improves the rate of dark adaptation in Rlbp1 knockout mice.
Choi, VW; Bigelow, CE; McGee, TL; Gujar, AN; Li, H; Hanks, SM; Vrouvlianis, J; Maker, M; Leehy, B; Zhang, Y; Aranda, J; Bounoutas, G; Demirs, JT; Yang, J; Ornberg, R; Wang, Y; Martin, W; Stout, KR; Argentieri, G; Grosenstein, P; Diaz, D; Turner, O; Jaffee, BD; Police, SR; Dryja, TP
Molecular therapy. Methods & clinical development
2
15022
2015
요약 표시
Recessive mutations in RLBP1 cause a form of retinitis pigmentosa in which the retina, before its degeneration leads to blindness, abnormally slowly recovers sensitivity after exposure to light. To develop a potential gene therapy for this condition, we tested multiple recombinant adeno-associated vectors (rAAVs) composed of different promoters, capsid serotypes, and genome conformations. We generated rAAVs in which sequences from the promoters of the human RLBP1, RPE65, or BEST1 genes drove the expression of a reporter gene (green fluorescent protein). A promoter derived from the RLBP1 gene mediated expression in the retinal pigment epithelium and Müller cells (the intended target cell types) at qualitatively higher levels than in other retinal cell types in wild-type mice and monkeys. With this promoter upstream of the coding sequence of the human RLBP1 gene, we compared the potencies of vectors with an AAV2 versus an AAV8 capsid in transducing mouse retinas, and we compared vectors with a self-complementary versus a single-stranded genome. The optimal vector (scAAV8-pRLBP1-hRLBP1) had serotype 8 capsid and a self-complementary genome. Subretinal injection of scAAV8-pRLBP1-hRLBP1 in Rlbp1 nullizygous mice improved the rate of dark adaptation based on scotopic (rod-plus-cone) and photopic (cone) electroretinograms (ERGs). The effect was still present after 1 year. | | | 26199951
|
Gene Therapy Fully Restores Vision to the All-Cone Nrl(-/-) Gucy2e(-/-) Mouse Model of Leber Congenital Amaurosis-1.
Boye, SL; Peterson, JJ; Choudhury, S; Min, SH; Ruan, Q; McCullough, KT; Zhang, Z; Olshevskaya, EV; Peshenko, IV; Hauswirth, WW; Ding, XQ; Dizhoor, AM; Boye, SE
Human gene therapy
26
575-92
2015
요약 표시
Mutations in GUCY2D are the cause of Leber congenital amaurosis type 1 (LCA1). GUCY2D encodes retinal guanylate cyclase-1 (retGC1), a protein expressed exclusively in outer segments of photoreceptors and essential for timely recovery from photoexcitation. Recent clinical data show that, despite a high degree of visual disturbance stemming from a loss of cone function, LCA1 patients retain normal photoreceptor architecture, except for foveal cone outer segment abnormalities and, in some patients, foveal cone loss. These results point to the cone-rich central retina as a target for GUCY2D replacement. LCA1 gene replacement studies thus far have been conducted in rod-dominant models (mouse) or with vectors and organisms lacking clinical translatability. Here we investigate gene replacement in the Nrl(-/-) Gucy2e(-/-) mouse, an all-cone model deficient in retGC1. We show that AAV-retGC1 treatment fully restores cone function, cone-mediated visual behavior, and guanylate cyclase activity, and preserves cones in treated Nrl(-/-) Gucy2e(-/-) mice over the long-term. A novel finding was that retinal function could be restored to levels above that in Nrl(-/-) controls, contrasting results in other models of retGC1 deficiency. We attribute this to increased cyclase activity in treated Nrl(-/-) Gucy2e(-/-) mice relative to Nrl(-/-) controls. Thus, Nrl(-/-) Gucy2e(-/-) mice possess an expanded dynamic range in ERG response to gene replacement relative to other models. Lastly, we show that a candidate clinical vector, AAV5-GRK1-GUCY2D, when delivered to adult Nrl(-/-) Gucy2e(-/-) mice, restores retinal function that persists for at least 6 months. Our results provide strong support for clinical application of a gene therapy targeted to the cone-rich, central retina of LCA1 patients. | | | 26247368
|
Long-term retinal cone survival and delayed alteration of the cone mosaic in a transgenic mouse model of stargardt-like dystrophy (STGD3).
Kuny, S; Filion, MA; Suh, M; Gaillard, F; Sauvé, Y
Investigative ophthalmology & visual science
55
424-39
2014
요약 표시
To examine the pattern of cone degeneration in the retina of a transgenic mouse model of Stargartd-like dystrophy (STGD3).Investigations were performed on ELOVL4/TG1-2 transgenic (TG) mice and wild-type (WT) littermates from 1 to 24 months of age. Phenotypes were assessed by fundus imaging, fatty acid analysis, and electroretinogram (ERG) recording. Cone degeneration pattern was determined on retina whole mounts using immunohistochemistry and Voronoi domain analyses.Consistent with low transgene expression, photoreceptors degenerate very slowly. At 1 month, anatomical structure and fatty acid composition of the TG retina is comparable with WT. Rod loss appears at 2 months, exhibiting a central to peripheral gradient, and fundus defects are observed at 3 months. In contrast, cone morphology, distribution and function are still normal at 12 months. Cone loss becomes apparent at 15 months when the outer nuclear layer is reduced to 3 to 4 photoreceptor rows. This process starts at the center of the retina and affects cone subtypes similarly. Very few cones remain at 24 months, after all rods have disappeared (18 months). Quantitative studies focusing on cones expressing M-opsin show a net increase in Voronoi domains and a significant decrease in regularity indexes only beyond 15 months.Photoreceptor degeneration in this STGD3 mouse model follows the time course of a slow rod-cone dystrophy. The cone mosaic is preserved for almost 1 year after the onset of rod loss. This long delay provides an opportunity to examine rod-cone interactions during retinal degeneration and to test therapeutic effectiveness at protracting cone dysfunction. | | | 24334447
|
Three distinct blue-green color pathways in a mammalian retina.
Mills, SL; Tian, LM; Hoshi, H; Whitaker, CM; Massey, SC
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
1760-8
2014
요약 표시
In mammalian retinae, the first steps in the process of discrimination of color are mediated by color-opponent neurons that respond with opposite polarity to signals from short (S, blue) and longer wavelength (M, green or L, red) cones. Primates also contain a second system that is different from M and L cones. Although pathways responding to the onset of S-cone stimulation (S-ON) are well known, the existence of bipolar cells and retinal ganglion cells that respond to the offset of S-cone stimulation (S-OFF) has been controversial. We have recorded from and stained three different types of S/M color-opponent ganglion cells in the rabbit retina that are distinguished by the polarity of their responses to S-cone stimulation, the stratification pattern of their dendrites, and the distinct mechanisms underlying their color-opponent responses. We describe an S-ON and an S-OFF pathway formed by amacrine cells inverting the S-ON signal. Most importantly, we also provide both anatomical and physiological evidence for a direct S-OFF pathway dependent on an S-OFF cone bipolar cell. The results indicate a greater diversity of pathways for processing of signals from S-cones than previously suspected. | | | 24478358
|
Natural history of cone disease in the murine model of Leber congenital amaurosis due to CEP290 mutation: determining the timing and expectation of therapy.
Boye, SE; Huang, WC; Roman, AJ; Sumaroka, A; Boye, SL; Ryals, RC; Olivares, MB; Ruan, Q; Tucker, BA; Stone, EM; Swaroop, A; Cideciyan, AV; Hauswirth, WW; Jacobson, SG
PloS one
9
e92928
2014
요약 표시
Mutations in the CEP290 (cilia-centrosomal protein 290 kDa) gene in Leber congenital amaurosis (LCA) cause early onset visual loss but retained cone photoreceptors in the fovea, which is the potential therapeutic target. A cone-only mouse model carrying a Cep290 gene mutation, rd16;Nrl-/-, was engineered to mimic the human disease. In the current study, we determined the natural history of retinal structure and function in this murine model to permit design of pre-clinical proof-of-concept studies and allow progress to be made toward human therapy. Analyses of retinal structure and visual function in CEP290-LCA patients were also performed for comparison with the results in the model.Rd16;Nrl-/- mice were studied in the first 90 days of life with optical coherence tomography (OCT), electroretinography (ERG), retinal histopathology and immunocytochemistry. Structure and function data from a cohort of patients with CEP290-LCA (n = 15; ages 7-48) were compared with those of the model.CEP290-LCA patients retain a central island of photoreceptors with normal thickness at the fovea (despite severe visual loss); the extent of this island declined slowly with age. The rd16;Nrl-/- model also showed a relatively slow photoreceptor layer decline in thickness with ∼80% remaining at 3 months. The number of pseudorosettes also became reduced. By comparison to single mutant Nrl-/- mice, UV- and M-cone ERGs of rd16;Nrl-/- were at least 1 log unit reduced at 1 month of age and declined further over the 3 months of monitoring. Expression of GNAT2 and S-opsin also decreased with age.The natural history of early loss of photoreceptor function with retained cone cell nuclei is common to both CEP290-LCA patients and the rd16;Nrl-/- murine model. Pre-clinical proof-of-concept studies for uniocular therapies would seem most appropriate to begin with intervention at P35-40 and re-study after one month by assaying interocular difference in the UV-cone ERG. | | | 24671090
|
Onecut1 and Onecut2 redundantly regulate early retinal cell fates during development.
Sapkota, D; Chintala, H; Wu, F; Fliesler, SJ; Hu, Z; Mu, X
Proceedings of the National Academy of Sciences of the United States of America
111
E4086-95
2014
요약 표시
Previously, we have shown that Onecut1 (Oc1) and Onecut2 (Oc2) are expressed in retinal progenitor cells, developing retinal ganglion cells (RGCs), and horizontal cells (HCs). However, in Oc1-null mice, we only observed an 80% reduction in HCs, but no defects in other cell types. We postulated that the lack of defects in other cell types in Oc1-null retinas was a result of redundancy with Oc2. To test this theory, we have generated Oc2-null mice and now show that their retinas also only have defects in HCs, with a 50% reduction in their numbers. However, when both Oc1 and Oc2 are knocked out, the retinas exhibit more profound defects in the development of all early retinal cell types, including completely failed genesis of HCs, compromised generation of cones, reduced production (by 30%) of RGCs, and absence of starburst amacrine cells. Cone subtype diversification and RGC subtype composition also were affected in the double-null retina. Using RNA-Seq expression profiling, we have identified downstream genes of Oc1 and Oc2, which not only confirms the redundancy between the two factors and renders a molecular explanation for the defects in the double-null retinas, but also shows that the onecut factors suppress the production of the late cell type, rods, indicating that the two factors contribute to the competence of retinal progenitor cells for the early retinal cell fates. Our results provide insight into how onecut factors regulate the creation of cellular diversity in the retina and, by extension, in the central nervous system in general. | | | 25228773
|
Independent genomic control of neuronal number across retinal cell types.
Keeley, PW; Whitney, IE; Madsen, NR; St John, AJ; Borhanian, S; Leong, SA; Williams, RW; Reese, BE
Developmental cell
30
103-9
2014
요약 표시
The sizes of different neuronal populations within the CNS are precisely controlled, but whether neuronal number is coordinated between cell types is unknown. We examined the covariance structure of 12 different retinal cell types across 30 genetically distinct lines of mice, finding minimal covariation when comparing synaptically connected or developmentally related cell types. Variation mapped to one or more genomic loci for each cell type, but rarely were these shared, indicating minimal genetic coregulation of final number. Multiple genes, therefore, participate in the specification of the size of every population of retinal neuron, yet genetic variants work largely independent of one another during development to modulate those numbers, yielding substantial variability in the convergence ratios between pre- and postsynaptic populations. Density-dependent cellular interactions in the outer plexiform layer overcome this variability to ensure the formation of neuronal circuits that maintain constant retinal coverage and complete afferent sampling. | | | 24954025
|