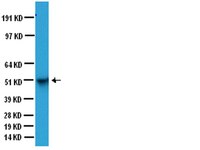
Endothelin-1 induces LIMK2-mediated programmed necrotic neuronal death independent of NOS activity.
Ko, AR; Hyun, HW; Min, SJ; Kim, JE; Kang, TC
Molecular brain
8
58
2015
요약 표시
Recently, we have reported that LIM kinase 2 (LIMK2) involves programmed necrotic neuronal deaths induced by aberrant cyclin D1 expression following status epilepticus (SE). Up-regulation of LIMK2 expression induces neuronal necrosis by impairment of dynamin-related protein 1 (DRP1)-mediated mitochondrial fission. However, we could not elucidate the upstream effecter for LIMK2-mediated neuronal death. Thus, we investigated the role of endothelin-1 (ET-1) in LIMK2-mediated neuronal necrosis, since ET-1 involves neuronal death via various pathways.Following SE, ET-1 concentration and its mRNA were significantly increased in the hippocampus with up-regulation of ETB receptor expression. BQ788 (an ETB receptor antagonist) effectively attenuated SE-induced neuronal damage as well as reduction in LIMK2 mRNA/protein expression. In addition, BQ788 alleviated up-regulation of Rho kinase 1 (ROCK1) expression and impairment of DRP1-mediated mitochondrial fission in CA1 neurons following SE. BQ788 also attenuated neuronal death and up-regulation of LIMK2 expression induced by exogenous ET-1 injection.These findings suggest that ET-1 may be one of the upstream effectors for programmed neuronal necrosis through abnormal LIMK2 over-expression by ROCK1. | | | 26438559
 |
Non-neuronal and neuronal BACE1 elevation in association with angiopathic and leptomeningeal β-amyloid deposition in the human brain.
Xue, ZQ; He, ZW; Yu, JJ; Cai, Y; Qiu, WY; Pan, A; Gai, WP; Cai, H; Luo, XG; Ma, C; Yan, XX
BMC neurology
15
71
2015
요약 표시
Cerebral amyloid angiopathy (CAA) refers to the deposition of β-amyloid (Aβ) peptides in the wall of brain vasculature, commonly involving capillaries and arterioles. Also being considered a part of CAA is the Aβ deposition in leptomeninge. The cellular origin of angiopathic Aβ and the pathogenic course of CAA remain incompletely understood.The present study was aimed to explore the pathogenic course of CAA in the human cerebrum via examination of changes in β-secretase-1 (BACE1), the obligatory Aβ producing enzyme, relative to Aβ and other cellular markers, by neuroanatomical and biochemical characterizations with postmortem brain samples and primary cell cultures.Immunoreactivity (IR) for BACE1 was essentially not visible at vasculature in cases without cerebral amyloidosis (control group, n = 15, age = 86.1 ± 10.3 year). In cases with brain amyloid pathology (n = 15, age = 78.7 ± 12.7 year), increased BACE1 IR was identified locally at capillaries, arterioles and along the pia, localizing to endothelia, perivascular dystrophic neurites and meningeal cells, and often coexisting with vascular iron deposition. Double immunofluorescence with densitometric analysis confirmed a site-specific BACE1 elevation at cerebral arterioles in the development of vascular Aβ deposition. Levels of BACE1 protein, activity and its immediate product (C99) were elevated in leptomeningeal lysates from cases with CAA relative to controls. The expression of BACE1 and other amyloidogenic proteins in the endothelial and meningeal cells was confirmed in primary cultures prepared from human leptomeningeal and arteriolar biopsies.These results suggest that BACE1 elevation in the endothelia and perivascular neurites may be involved in angiopathic Aβ deposition, while BACE1 elevation in meningeal cells might contribute Aβ to leptomeningeal amyloidosis. | | | 25934480
|
Netrin-5 is highly expressed in neurogenic regions of the adult brain.
Yamagishi, S; Yamada, K; Sawada, M; Nakano, S; Mori, N; Sawamoto, K; Sato, K
Frontiers in cellular neuroscience
9
146
2015
요약 표시
Mammalian netrin family proteins are involved in targeting of axons, neuronal migration, and angiogenesis and act as repulsive and attractive guidance molecules. Netrin-5 is a new member of the netrin family with homology to the C345C domain of netrin-1. Unlike other netrin proteins, murine netrin-5 consists of two EGF motifs of the laminin V domain (LE) and the C345C domain, but lacks the N-terminal laminin VI domain and one of the three LE motifs. We generated a specific antibody against netrin-5 to investigate its expression pattern in the rodent adult brain. Strong netrin-5 expression was observed in the olfactory bulb (OB), rostral migrate stream (RMS), the subventricular zone (SVZ), and the subgranular zone (SGZ) of the dentate gyrus in the hippocampus, where neurogenesis occurs in the adult brain. In the SVZ and RMS, netrin-5 expression was observed in Mash1-positive transit-amplifying cells and in Doublecortin (DCX)-positive neuroblasts, but not in GFAP-positive astrocytes. In the OB, netrin-5 expression was maintained in neuroblasts, but its level was decreased in NeuN-positive mature neurons. In the hippocampal SGZ, netrin-5 was observed in Mash1-positive cells and in DCX-positive neuroblasts, but not in GFAP-positive astrocytes, suggesting that netrin-5 expression occurs from type 2a to type 3 cells. These data suggest that netrin-5 is produced by both transit-amplifying cells and neuroblasts to control neurogenesis in the adult brain. | | | 25941474
|
Involvement of phosphatase and tensin homolog deleted from chromosome 10 in rodent model of neuropathic pain.
Huang, SY; Sung, CS; Chen, WF; Chen, CH; Feng, CW; Yang, SN; Hung, HC; Chen, NF; Lin, PR; Chen, SC; Wang, HM; Chu, TH; Tai, MH; Wen, ZH
Journal of neuroinflammation
12
59
2015
요약 표시
Many cancer research studies have extensively examined the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) pathway. There are only few reports that suggest that PTEN might affect pain; however, there is still a lack of evidence to show the role of PTEN for modulating pain. Here, we report a role for PTEN in a rodent model of neuropathic pain.We found that chronic constriction injury (CCI) surgery in rats could elicit downregulation of spinal PTEN as well as upregulation of phosphorylated PTEN (phospho-PTEN) and phosphorylated mammalian target of rapamycin (phospho-mTOR). After examining such changes in endogenous PTEN in neuropathic rats, we explored the effects of modulating the spinal PTEN pathway on nociceptive behaviors. The normal rats exhibited mechanical allodynia after intrathecal (i.t.) injection of adenovirus-mediated PTEN antisense oligonucleotide (Ad-antisense PTEN). These data indicate the importance of downregulation of spinal PTEN for nociception. Moreover, upregulation of spinal PTEN by i.t. adenovirus-mediated PTEN (Ad-PTEN) significantly prevented CCI-induced development of nociceptive sensitization, thermal hyperalgesia, mechanical allodynia, cold allodynia, and weight-bearing deficits in neuropathic rats. Furthermore, upregulation of spinal PTEN by i.t. Ad-PTEN significantly attenuated CCI-induced microglia and astrocyte activation, upregulation of tumor necrosis factor-α (TNF-α) and phospho-mTOR, and downregulation of PTEN in neuropathic rats 14 days post injury.These findings demonstrate that PTEN plays a key, beneficial role in a rodent model of neuropathic pain. | | | 25889774
|
Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory.
Orr, AG; Hsiao, EC; Wang, MM; Ho, K; Kim, DH; Wang, X; Guo, W; Kang, J; Yu, GQ; Adame, A; Devidze, N; Dubal, DB; Masliah, E; Conklin, BR; Mucke, L
Nature neuroscience
18
423-34
2015
요약 표시
Astrocytes express a variety of G protein-coupled receptors and might influence cognitive functions, such as learning and memory. However, the roles of astrocytic Gs-coupled receptors in cognitive function are not known. We found that humans with Alzheimer's disease (AD) had increased levels of the Gs-coupled adenosine receptor A2A in astrocytes. Conditional genetic removal of these receptors enhanced long-term memory in young and aging mice and increased the levels of Arc (also known as Arg3.1), an immediate-early gene that is required for long-term memory. Chemogenetic activation of astrocytic Gs-coupled signaling reduced long-term memory in mice without affecting learning. Like humans with AD, aging mice expressing human amyloid precursor protein (hAPP) showed increased levels of astrocytic A2A receptors. Conditional genetic removal of these receptors enhanced memory in aging hAPP mice. Together, these findings establish a regulatory role for astrocytic Gs-coupled receptors in memory and suggest that AD-linked increases in astrocytic A2A receptor levels contribute to memory loss. | | | 25622143
|
Localization of reelin signaling pathway components in murine midbrain and striatum.
Sharaf, A; Rahhal, B; Spittau, B; Roussa, E
Cell and tissue research
359
393-407
2015
요약 표시
We investigated the distribution patterns of the extracellular matrix protein Reelin and of crucial Reelin signaling components in murine midbrain and striatum. The cellular distribution of the Reelin receptors VLDLr and ApoER2, the intracellular downstream mediator Dab1, and the alternative Reelin receptor APP were analyzed at embryonic day 16, at postnatal stage 15 (P15), and in 3-month-old mice. Reelin was expressed intracellularly and extracellularly in midbrain mesencephalic dopaminergic (mDA) neurons of newborns. In the striatum, Calbindin D-28k(+) neurons exhibited Reelin intracellularly at E16 and extracellularly at P15 and 3 months. ApoER2 and VLDLr were expressed in mDA neurons at E16 and P15 and in oligodendrocytes at 3 months, whereas Dab1 and APP immunoreactivity was observed in mDA at all stages analyzed. In the striatum, Calbindin D-28k(+)/GAD67(+) inhibitory neurons expressed VLDLr, ApoER2, and Dab1 at P15, but only Dab1 at E16 and 3 months. APP was always expressed in mouse striatum in which it colocalized with Calbindin D-28k. Our data underline the importance of Reelin signalling during embryonic development and early postnatal maturation of the mesostriatal and mesocorticolimbic system, and suggest that the striatum and not the midbrain is the primary source of Reelin for midbrain neurons. The loss of ApoER2 and VLDLr expression in the mature midbrain and striatum implies that Reelin functions are restricted to migratory events and early postnatal maturation and are dispensable for the maintenance of dopaminergic neurons. | | Mouse | 25418135
|
Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice.
Baratz, R; Tweedie, D; Wang, JY; Rubovitch, V; Luo, W; Hoffer, BJ; Greig, NH; Pick, CG
Journal of neuroinflammation
12
45
2015
요약 표시
The treatment of traumatic brain injury (TBI) represents an unmet medical need, as no effective pharmacological treatment currently exists. The development of such a treatment requires a fundamental understanding of the pathophysiological mechanisms that underpin the sequelae resulting from TBI, particularly the ensuing neuronal cell death and cognitive impairments. Tumor necrosis factor-alpha (TNF-α) is a cytokine that is a master regulator of systemic and neuroinflammatory processes. TNF-α levels are reported to become rapidly elevated post TBI and, potentially, can lead to secondary neuronal damage.To elucidate the role of TNF-α in TBI, particularly as a drug target, the present study evaluated (i) time-dependent TNF-α levels and (ii) markers of apoptosis and gliosis within the brain and related these to behavioral measures of 'well being' and cognition in a mouse closed head 50 g weight drop mild TBI (mTBI) model in the presence and absence of post-treatment with an experimental TNF-α synthesis inhibitor, 3,6'-dithiothalidomide.mTBI elevated brain TNF-α levels, which peaked at 12 h post injury and returned to baseline by 18 h. This was accompanied by a neuronal loss and an increase in astrocyte number (evaluated by neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) immunostaining), as well as an elevation in the apoptotic death marker BH3-interacting domain death agonist (BID) at 72 h. Selective impairments in measures of cognition, evaluated by novel object recognition and passive avoidance paradigms - without changes in well being, were evident at 7 days after injury. A single systemic treatment with the TNF-α synthesis inhibitor 3,6'-dithiothalidomide 1 h post injury prevented the mTBI-induced TNF-α elevation and fully ameliorated the neuronal loss (NeuN), elevations in astrocyte number (GFAP) and BID, and cognitive impairments. Cognitive impairments evident at 7 days after injury were prevented by treatment as late as 12 h post mTBI but were not reversed when treatment was delayed until 18 h.These results implicate that TNF-α in mTBI induced secondary brain damage and indicate that pharmacologically limiting the generation of TNF-α post mTBI may mitigate such damage, defining a time-dependent window of up to 12 h to achieve this reversal. | | | 25879458
|
Human Embryonic Stem Cell-Derived Progenitors Assist Functional Sensory Axon Regeneration after Dorsal Root Avulsion Injury.
Hoeber, J; Trolle, C; Konig, N; Du, Z; Gallo, A; Hermans, E; Aldskogius, H; Shortland, P; Zhang, SC; Deumens, R; Kozlova, EN
Scientific reports
5
10666
2015
요약 표시
Dorsal root avulsion results in permanent impairment of sensory functions due to disconnection between the peripheral and central nervous system. Improved strategies are therefore needed to reconnect injured sensory neurons with their spinal cord targets in order to achieve functional repair after brachial and lumbosacral plexus avulsion injuries. Here, we show that sensory functions can be restored in the adult mouse if avulsed sensory fibers are bridged with the spinal cord by human neural progenitor (hNP) transplants. Responses to peripheral mechanical sensory stimulation were significantly improved in transplanted animals. Transganglionic tracing showed host sensory axons only in the spinal cord dorsal horn of treated animals. Immunohistochemical analysis confirmed that sensory fibers had grown through the bridge and showed robust survival and differentiation of the transplants. Section of the repaired dorsal roots distal to the transplant completely abolished the behavioral improvement. This demonstrates that hNP transplants promote recovery of sensorimotor functions after dorsal root avulsion, and that these effects are mediated by spinal ingrowth of host sensory axons. These results provide a rationale for the development of novel stem cell-based strategies for functionally useful bridging of the peripheral and central nervous system. | Immunohistochemistry | | 26053681
|
Long-term vision and non-vision dominant behavioral deficits in the 2-VO rats are accompanied by time and regional glial activation in the white matter.
Tian, XS; Guo, XJ; Ruan, Z; Lei, Y; Chen, YT; Zhang, HY
PloS one
9
e101120
2014
요약 표시
The permanent occlusion of common carotid arteries (2-VO) in rats has been shown to induce progressive and long-lasting deficits in cognitive performance, however, whether these aberrant behaviors are attributed to visual dysfunction or cognitive impairment and what are the underlying mechanisms, remain controversial. In the present study, vision dominant (Morris water maze) and non-vision dominant (voice-cued fear conditioning) behavioral tests were assigned to comprehensively evaluate the influence of 2-VO lesion on cognitive behaviors. In the Morris water maze test, escape latencies of 2-VO rats were markedly increased in both hidden and unfixed visible platform tasks, which were accompanied by severe retinal damage. In the voice-cued fear conditioning test, significant reduction in the percentage of freezing behavior was observed at 60 days after 2-VO lesion. Chronic lesion by 2-VO failed to cause noticeable changes in the grey matter, as indicated by intact hippocampal and prefrontal cortical structures, sustained synaptic protein levels and glial cell numbers. In contrast, aberrant arrangement of myelinated axons was observed in the optic tract, but not in the corpus callosum and inner capsule of 2-VO rats. Concurrently, marked astrocyte proliferation and microglia activation in the optic tract occurred at 3 days after 2-VO lesion, and continued for up to 60 days. Differently, robust glial activation was observed in the corpus callosum at 3 days after 2-VO surgery, and then gradually returned to the baseline level at 14 and 60 days. Our study reported for the first time about the effect of 2-VO on the long-term cognitive impairment in the non-vision dominant fear conditioning test, which may be more applicable than the Morris water maze test for assessing 2-VO associated cognitive function. The time and region specific glial activation in the white matter may relate to retinal impairment, even behavioral deficits, in the setting of chronic cerebral hypoperfusion. | | | 24968196
|
Hepatic stellate cells contribute to progenitor cells and liver regeneration.
Kordes, C; Sawitza, I; Götze, S; Herebian, D; Häussinger, D
The Journal of clinical investigation
124
5503-15
2014
요약 표시
Retinoid-storing hepatic stellate cells (HSCs) have recently been described as a liver-resident mesenchymal stem cell (MSC) population; however, it is not clear whether these cells contribute to liver regeneration or serve as a progenitor cell population with hepatobiliary characteristics. Here, we purified HSCs with retinoid-dependent fluorescence-activated cell sorting from eGFP-expressing rats and transplanted these GFP(+) HSCs into wild-type (WT) rats that had undergone partial hepatectomy in the presence of 2-acetylaminofluorene (2AAF) or retrorsine, both of which are injury models that favor stem cell-based liver repair. Transplanted HSCs contributed to liver regeneration in host animals by forming mesenchymal tissue, progenitor cells, hepatocytes, and cholangiocytes and elevated direct bilirubin levels in blood sera of GUNN rats, indicating recovery from the hepatic bilirubin-handling defect in these animals. Transplanted HSCs engrafted within the bone marrow (BM) of host animals, and HSC-derived cells were isolated from BM and successfully retransplanted into new hosts with injured liver. Cultured HSCs transiently adopted an expression profile similar to that of progenitor cells during differentiation into bile acid-synthesizing and -transporting hepatocytes, suggesting that stellate cells represent a source of liver progenitor cells. This concept connects seemingly contradictory studies that favor either progenitor cells or MSCs as important players in stem cell-based liver regeneration. | | | 25401473
|