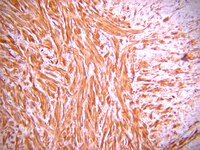
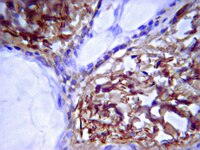
Recessive COL6A2 C-globular missense mutations in Ullrich congenital muscular dystrophy: role of the C2a splice variant.
Zhang RZ, Zou Y, Pan TC, Markova D, Fertala A, Hu Y, Squarzoni S, Reed UC, Marie SK, Bönnemann CG, Chu ML
J Biol Chem
285
10005-15 Epub 2010 Jan 27
2010
요약 표시
Ullrich congenital muscular dystrophy (UCMD) is a disabling and life-threatening disorder resulting from either recessive or dominant mutations in genes encoding collagen VI. Although the majority of the recessive UCMD cases have frameshift or nonsense mutations in COL6A1, COL6A2, or COL6A3, recessive structural mutations in the COL6A2 C-globular region are emerging also. However, the underlying molecular mechanisms have remained elusive. Here we identified a homozygous COL6A2 E624K mutation (C1 subdomain) and a homozygous COL6A2 R876S mutation (C2 subdomain) in two UCMD patients. The consequences of the mutations were investigated using fibroblasts from patients and cells stably transfected with the mutant constructs. In contrast to expectations based on the clinical severity of these two patients, secretion and assembly of collagen VI were moderately affected by the E624K mutation but severely impaired by the R876S substitution. The E624K substitution altered the electrostatic potential of the region surrounding the metal ion-dependent adhesion site, resulting in a collagen VI network containing thick fibrils and spots with densely packed microfibrils. The R876S mutation prevented the chain from assembling into triple-helical collagen VI molecules. The minute amount of collagen VI secreted by the R876S fibroblasts was solely composed of a faster migrating chain corresponding to the C2a splice variant with an alternative C2 subdomain. In transfected cells, the C2a splice variant was able to assemble into short microfibrils. Together, the results suggest that the C2a splice variant may functionally compensate for the loss of the normal COL6A2 chain when mutations occur in the C2 subdomain. 기사 전문 | 20106987
 |
Predominant fiber atrophy and fiber type disproportion in early ullrich disease.
Joachim Schessl, Nathalie M Goemans, Alexandra I Magold, Yaqun Zou, Ying Hu, Janbernd Kirschner, Raf Sciot, Carsten G Bönnemann
Muscle nerve
38
1184-91
2008
요약 표시
Ullrich disease (congenital muscular dystrophy type Ullrich, UCMD) is a severe congenital disorder of muscle caused by recessive and dominant mutations in the three genes that encode the alpha-chains of collagen type VI. Little is known about the early pathogenesis of this myopathy. The aim of this study was to investigate early histological changes in muscle of patients with molecularly confirmed UCMD. Muscle biopsies were analyzed from 8 UCMD patients ranging in age from 6 to 30 months. Type I fiber atrophy and predominance were seen early, together with a widening of the fiber diameter spectrum, whereas no dystrophic features were apparent. A subpopulation of more severely atrophic type I fibers was apparent subsequently, including one biopsy that fulfilled the formal diagnostic criteria of histopathological fiber type disproportion (FTD). Thus, early in the disease, UCMD presents as a non-dystrophic myopathy with predominant fiber atrophy. Collagen VI mutations also qualify as a cause of fiber type disproportion. | 18720506
|
Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance.
A K Lampe,Y Zou,D Sudano,K K O'Brien,D Hicks,S H Laval,R Charlton,C Jimenez-Mallebrera,R-Z Zhang,R S Finkel,G Tennekoon,G Schreiber,M S van der Knaap,H Marks,V Straub,K M Flanigan,M-L Chu,F Muntoni,K M D Bushby,C G Bönnemann
Human mutation
29
2008
요약 표시
Mutations in the genes encoding collagen VI (COL6A1, COL6A2, and COL6A3) cause Bethlem myopathy (BM) and Ullrich congenital muscular dystrophy (UCMD), two related conditions of differing severity. BM is a relatively mild dominantly inherited disorder characterized by proximal weakness and distal joint contractures. UCMD was originally regarded as an exclusively autosomal recessive condition causing severe muscle weakness with proximal joint contractures and distal hyperlaxity. We and others have subsequently modified this model when we described UCMD patients with heterozygous in-frame deletions acting in a dominant-negative way. Here we report 10 unrelated patients with a UCMD clinical phenotype and de novo dominant negative heterozygous splice mutations in COL6A1, COL6A2, and COL6A3 and contrast our findings with four UCMD patients with recessively acting splice mutations and two BM patients with heterozygous splice mutations. We find that the location of the skipped exon relative to the molecular structure of the collagen chain strongly correlates with the clinical phenotype. Analysis by immunohistochemical staining of muscle biopsies and dermal fibroblast cultures, as well as immunoprecipitation to study protein biosynthesis and assembly, suggests different mechanisms each for exon skipping mutations underlying dominant UCMD, dominant BM, and recessive UCMD. We provide further evidence that de novo dominant mutations in severe UCMD occur relatively frequently in all three collagen VI chains and offer biochemical insight into genotype-phenotype correlations within the collagen VI-related disorders by showing that severity of the phenotype depends on the ability of mutant chains to be incorporated in the multimeric structure of collagen VI. | 18366090
|
A new form of congenital muscular dystrophy with joint hyperlaxity maps to 3p23-21.
Tétreault, M; Duquette, A; Thiffault, I; Bherer, C; Jarry, J; Loisel, L; Banwell, B; D'Anjou, G; Mathieu, J; Robitaille, Y; Vanasse, M; Brais, B
Brain : a journal of neurology
129
2077-84
2006
요약 표시
Congenital muscular dystrophies (CMDS) are a heterogeneous group of disorders. A growing number of CMDS have been found to be associated with joint hyperlaxity. We recruited 14 French-Canadian cases belonging to 11 families affected by a novel autosomal recessive congenital muscular dystrophy with hyperlaxity (CMDH). All cases come from the southwestern part of Quebec, suggesting a new French-Canadian founder effect. All patients present muscle weakness, proximal contractures coexisting with distal joint hyperlaxity. Pathological and genetic studies have excluded that mutations in the three genes coding for collagen VI subunits are responsible for this disease. A genome-wide scan established linkage of two CMDH families to a region on chromosome 3p23-21. Further linkage analysis confirmed that all families are linked to the same region (log of the odds score of 5.3). Haplotype analysis defines a 1.6-cM candidate interval and suggests that two common mutations may account for 78% of carrier chromosomes. This study describes and maps a new form of recessive CMD with joint hyperlaxity distinct from Ullrich and Bethlem myopathies with a founder effect in the French-Canadian population. | 16760198
|
Ullrich congenital muscular dystrophy: connective tissue abnormalities in the skin support overlap with Ehlers-Danlos syndromes.
Janbernd Kirschner, Ingrid Hausser, Yaqun Zou, Gudrun Schreiber, Hans-Jürgen Christen, Susan C Brown, Ingrun Anton-Lamprecht, Francesco Muntoni, Folker Hanefeld, Carsten G Bönnemann
American journal of medical genetics. Part A
132A
296-301
2005
요약 표시
Ullrich congenital muscular dystrophy (UCMD) is caused by mutations in the three genes coding for the alpha chains of collagen VI and characterized by generalized muscle weakness, striking hypermobility of distal joints in conjunction with variable contractures of more proximal joints, and normal intellectual development. The diagnosis is supported by abnormal immunoreactivity for collagen VI on muscle biopsies. As patients with UCMD show clinical characteristics typical of classical disorders of connective tissue such as Ehlers-Danlos syndromes (EDS), we investigated the ultrastructure of skin biopsy samples from patients with UCMD (n=5). Electron microscopy of skin biopsies revealed ultrastructural abnormalities in all cases, including alterations of collagen fibril morphology (variation in size and composite fibers) and increase in ground substance, which resemble those seen in patients with EDS. Our findings suggest that there is a true connective tissue component as part of the phenotypic spectrum of UCMD and that there is considerable clinical as well as morphological overlap between UCMD and classic connective tissue disorders. | 15690374
|
Kidneys with heavy proteinuria show fibrosis, inflammation, and oxidative stress, but no tubular phenotypic change.
Kuusniemi, AM; Lapatto, R; Holmberg, C; Karikoski, R; Rapola, J; Jalanko, H
Kidney international
68
121-32
2005
요약 표시
Sustained proteinuria is a major factor leading to kidney fibrosis and end-stage renal failure. Tubular epithelial cells are believed to play a crucial role in this process by producing mediators leading to fibrosis and inflammation. Congenital nephrotic syndrome of the Finnish type (NPHS1) is a genetic disease caused by mutations in a podocyte protein nephrin, which leads to constant heavy proteinuria from birth. In this work we studied the tubulointerstitial changes that occur in NPHS1 kidneys during infancy.The pathologic lesions and expression of profibrotic and proinflammatory factors in nephrectomized NPHS1 kidneys were studied by immunohistochemistry, Western blotting, and cytokine antibody array. Oxidative stress in kidneys was assessed by measurement of gluthatione redox state.The results indicated that (1) severe tubulointerstitial lesions developed in NPHS1 kidneys during infancy; (2) tubular epithelial cells did not show transition into myofibroblasts as studied by the expression of vimentin, alpha-smooth muscle actin (alpha-SMA), collagen, and matrix metalloproteinases 2 and 9 (MMP-2 and -9); (3) the most abundant chemokines in NPHS1 tissue were neutrophil activating protein-2 (NAP-2), macrophage inhibiting factor (MIF), and monocyte chemoattractant protein-1 (MCP-1); (4) monocyte/macrophage cells expressing CD14 antigen were the major inflammatory cells invading the interstitium; (5) the arteries and arterioles showed intimal hypertrophy, but the microvasculature in NPHS1 kidneys remained quite normal; and (6) excessive oxidative stress was evident in NPHS1 kidneys.Heavy proteinuria in NPHS1 kidneys was associated with interstitial fibrosis, inflammation, and oxidative stress. The tubular epithelial cells, however, were resistant to proteinuria and did not show epithelial-mesenchymal transition. | 15954901
|
Collagen VI related muscle disorders.
Lampe, AK; Bushby, KM
Journal of medical genetics
42
673-85
2005
요약 표시
Mutations in the genes encoding collagen VI (COL6A1, COL6A2, and COL6A3) cause Bethlem myopathy (BM) and Ullrich congenital muscular dystrophy (UCMD), two conditions which were previously believed to be completely separate entities. BM is a relatively mild dominantly inherited disorder characterised by proximal weakness and distal joint contractures. UCMD was originally described as an autosomal recessive condition causing severe muscle weakness with proximal joint contractures and distal hyperlaxity. Here we review the clinical phenotypes of BM and UCMD and their diagnosis and management, and provide an overview of the current knowledge of the pathogenesis of collagen VI related disorders. 기사 전문 | 16141002
|
Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: phenotype analysis of 11 families linked to the COL6 loci.
E Demir, A Ferreiro, P Sabatelli, V Allamand, S Makri, B Echenne, M Maraldi, L Merlini, H Topaloglu, P Guicheney
Neuropediatrics
35
103-12
2004
요약 표시
Ullrich's congenital muscular dystrophy (UCMD) is an autosomal recessive myopathy characterised by neonatal muscle weakness, proximal joint contractures and distal hyperlaxity. Mutations in the COL6A1, COL6A2 (21 q22.3) and COL6A3 (2 q37) genes, encoding the alpha 1, alpha 2 and alpha 3 chains of collagen VI, respectively, have been recently identified as responsible for UCMD in a total of 9 families. We investigated in detail the clinical and morphological phenotype of 15 UCMD patients from 11 consanguineous families showing potential linkage either to 21 q22.3 (6 families) or to 2 q37 (5 families). Collagen VI deficiency was confirmed on muscle biopsies or skin fibroblasts in 8 families. Although all patients shared a common phenotype, a great variability in severity was observed. Collagen VI deficiency in muscle or cultured fibroblasts was complete in the severe cases and partial in the milder ones, which suggests a correlation between the degree of collagen VI deficiency and the clinical severity in UCMD. No significant phenotypical differences were found between the families linked to each of the 2 loci, which confirms UCMD as a unique entity with underlying genetic heterogeneity. | 15127309
|
Novel COL6A1 splicing mutation in a family affected by mild Bethlem myopathy.
Olga Camacho Vanegas, Rui-Zhu Zhang, Patrizia Sabatelli, Giovanna Lattanzi, Paola Bencivenga, Betti Giusti, Marta Columbaro, Mon-Li Chu, Luciano Merlini, Guglielmina Pepe
Muscle nerve
25
513-9
2002
요약 표시
Bethlem myopathy is an early-onset benign myopathy characterized by proximal muscular weakness and multiple flexion contractures. It is a dominantly inherited disorder associated with mutations in the three COL6 genes encoding type VI collagen. We detected a g-->a substitution at +1 position of COL6A1 intron 3 in a four-generation Italian family affected by a mild form of Bethlem myopathy. The mutation results in the activation of a cryptic splice donor site at the 3' end of exon 3, leading to the loss of 66 nucleotides and an in-frame deletion of 22 amino acids in the NH2-domain. Molecular analysis on fibroblasts of the propositus showed that the mutated mRNA was present and stable, but the mutated protein could not be detected. Western blot and immunofluorescence analyses showed a decreased level of collagen VI synthesis and deposition in fibroblasts of the propositus. Together, the results suggest that the mutated protein was highly unstable and rapidly degraded, and that the mild phenotype was caused by a reduced amount of normal collagen VI microfibrils. In addition, we demonstrated that lymphocytes can be used for the first mutation screening analysis of patients with Bethlem myopathy. | 11932968
|
Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy.
Demir, E; Sabatelli, P; Allamand, V; Ferreiro, A; Moghadaszadeh, B; Makrelouf, M; Topaloglu, H; Echenne, B; Merlini, L; Guicheney, P
American journal of human genetics
70
1446-58
2002
요약 표시
Ullrich congenital muscular dystrophy (UCMD) is an autosomal recessive disorder characterized by generalized muscular weakness, contractures of multiple joints, and distal hyperextensibility. Homozygous and compound heterozygous mutations of COL6A2 on chromosome 21q22 have recently been shown to cause UCMD. We performed a genomewide screening with microsatellite markers in a consanguineous family with three sibs affected with UCMD. Linkage of the disease to chromosome 2q37 was found in this family and in two others. We analyzed COL6A3, which encodes the alpha3 chain of collagen VI, and identified one homozygous mutation per family. In family I, the three sibs carried an A--greater than G transition in the splice-donor site of intron 29 (6930+5A--greater than G), leading to the skipping of exon 29, a partial reduction of collagen VI in muscle biopsy, and an intermediate phenotype. In family II, the patient had an unusual mild phenotype, despite a nonsense mutation, R465X, in exon 5. Analysis of the patient's COL6A3 transcripts showed the presence of various mRNA species-one of which lacked several exons, including the exon containing the nonsense mutation. The deleted splice variant encodes collagen molecules that have a shorter N-terminal domain but that may assemble with other chains and retain a functional role. This could explain the mild phenotype of the patient who was still ambulant at age 18 years and who showed an unusual combination of hyperlaxity and finger contractures. In family III, the patient had a nonsense mutation, R2342X, causing absence of collagen VI in muscle and fibroblasts, and a severe phenotype, as has been described in patients with UCMD. Mutations in COL6A3 are described in UCMD for the first time and illustrate the wide spectrum of phenotypes which can be caused by collagen VI deficiency. | 11992252
|