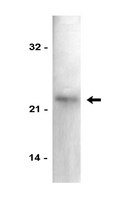
Identification and characterization of nuclear and nucleolar localization signals in 58-kDa microspherule protein (MSP58).
Yang, CP; Chiang, CW; Chen, CH; Lee, YC; Wu, MH; Tsou, YH; Yang, YS; Chang, WC; Lin, DY
Journal of biomedical science
22
33
2015
概要を表示する
MSP58 is a nucleolar protein associated with rRNA transcription and cell proliferation. Its mechanism of translocation into the nucleus or the nucleolus, however, is not entirely known. In order to address this lack, the present study aims to determine a crucial part of this mechanism: the nuclear localization signal (NLS) and the nucleolar localization signal (NoLS) associated with the MSP58 protein.We have identified and characterized two NLSs in MSP58. The first is located between residues 32 and 56 (NLS1) and constitutes three clusters of basic amino acids (KRASSQALGTIPKRRSSSRFIKRKK); the second is situated between residues 113 and 123 (NLS2) and harbors a monopartite signal (PGLTKRVKKSK). Both NLS1 and NLS2 are highly conserved among different vertebrate species. Notably, one bipartite motif within the NLS1 (residues 44-56) appears to be absolutely necessary for MSP58 nucleolar localization. By yeast two-hybrid, pull-down, and coimmunoprecipitation analysis, we show that MSP58 binds to importin α1 and α6, suggesting that nuclear targeting of MSP58 utilizes a receptor-mediated and energy-dependent import mechanism. Functionally, our data show that both nuclear and nucleolar localization of MSP58 are crucial for transcriptional regulation on p21 and ribosomal RNA genes, and context-dependent effects on cell proliferation.Results suggest that MSP58 subnuclear localization is regulated by two nuclear import signals, and that proper subcellular localization of MSP58 is critical for its role in transcriptional regulation. Our study reveals a molecular mechanism that controls nuclear and nucleolar localization of MSP58, a finding that might help future researchers understand the MSP58 biological signaling pathway. | | 25981436
 |
Gene profiling characteristics of radioadaptive response in AG01522 normal human fibroblasts.
Hou, J; Wang, F; Kong, P; Yu, PK; Wang, H; Han, W
PloS one
10
e0123316
2015
概要を表示する
Radioadaptive response (RAR) in mammalian cells refers to the phenomenon where a low-dose ionizing irradiation alters the gene expression profiles, and protects the cells from the detrimental effects of a subsequent high dose exposure. Despite the completion of numerous experimental studies on RAR, the underlying mechanism has remained unclear. In this study, we aimed to have a comprehensive investigation on the RAR induced in the AG01522 human fibroblasts first exposed to 5 cGy (priming dose) and then followed by 2 Gy (challenge dose) of X-ray through comparisons to those cells that had only received a single 2 Gy dose. We studied how the priming dose affected the expression of gene transcripts, and to identify transcripts or pathways that were associated with the reduced chromosomal damages (in terms of the number of micronuclei) after application of the challenging dose. Through the mRNA and microRNA microarray analyses, the transcriptome alteration in AG01522 cells was examined, and the significantly altered genes were identified for different irradiation procedures using bioinformatics approaches. We observed that a low-dose X-ray exposure produced an alert, triggering and altering cellular responses to defend against subsequent high dose-induced damages, and accelerating the cell repair process. Moreover, the p53 signaling pathway was found to play critial roles in regulating DNA damage responses at the early stage after application of the challenging dose, particularly in the RAR group. Furthermore, microRNA analyses also revealed that cell communication and intercellular signaling transduction played important roles after low-dose irradiation. We conclude that RAR benefits from the alarm mechanisms triggered by a low-dose priming radation dose. | | 25886619
|
High levels of TopBP1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation.
Sokka, M; Rilla, K; Miinalainen, I; Pospiech, H; Syväoja, JE
Nucleic acids research
43
4975-89
2015
概要を表示する
Nucleoli are not only organelles that produce ribosomal subunits. They are also overarching sensors of different stress conditions and they control specific nucleolar stress pathways leading to stabilization of p53. During DNA replication, ATR and its activator TopBP1 initiate DNA damage response upon DNA damage and replication stress. We found that a basal level of TopBP1 protein associates with ribosomal DNA repeat. When upregulated, TopBP1 concentrates at the ribosomal chromatin and initiates segregation of nucleolar components--the hallmark of nucleolar stress response. TopBP1-induced nucleolar segregation is coupled to shut-down of ribosomal RNA transcription in an ATR-dependent manner. Nucleolar segregation induced by TopBP1 leads to a moderate elevation of p53 protein levels and to localization of activated p53 to nucleolar caps containing TopBP1, UBF and RNA polymerase I. Our findings demonstrate that TopBP1 and ATR are able to inhibit the synthesis of rRNA and to activate nucleolar stress pathway; yet the p53-mediated cell cycle arrest is thwarted in cells expressing high levels of TopBP1. We suggest that inhibition of rRNA transcription by different stress regulators is a general mechanism for cells to initiate nucleolar stress pathway. | | 25916852
|
Loss of compensatory pro-survival and anti-apoptotic modulator, IKKε, sensitizes ovarian cancer cells to CHEK1 loss through an increased level of p21.
Kim, MK; Min, DJ; Wright, G; Goldlust, I; Annunziata, CM
Oncotarget
5
12788-802
2014
概要を表示する
Ovarian cancer (OC) is extremely heterogeneous, implying that therapeutic strategies should be specifically designed based on molecular characteristics of an individual's tumor. Previously, we showed that IKKε promotes invasion and metastasis in a subset of OCs. Here, we identified CHEK1 as an IKKε-dependent lethal gene from shRNA kinome library screen. In subsequent pharmacological intervention studies, the co-inhibition of IKKε and CHEK1 was more effective in killing OC cells than single treatment. At the molecular level, co-inhibition dramatically decreased pro-survival proteins, but increased proteins involved in DNA damage and apoptosis. IKKε-knockdown increased p21 levels, while overexpression of wild-type IKKε, but not a kinase dead IKKε mutant decreased p21 levels. We further demonstrated that the depletion of p21 rendered OC cells more resistant to cell death induced by co-inhibition of IKKε and CHEK1. In conclusion, we revealed a novel interplay between IKKε, CHEK1 and p21 signaling in survival of OC. Our study provides a rationale for the clinical development of specific IKKε inhibitor and for usage of IKKε as an exploratory marker for resistance to CHEK1 inhibitors in the clinic. The interplay provides one potential explanation as to why very few clinical responses were achieved in patients treated with single-agent CHEK1 inhibitors. | Western Blotting | 25474241
|
The downregulation of GFI1 by the EZH2-NDY1/KDM2B-JARID2 axis and by human cytomegalovirus (HCMV) associated factors allows the activation of the HCMV major IE promoter and the transition to productive infection.
Sourvinos, G; Morou, A; Sanidas, I; Codruta, I; Ezell, SA; Doxaki, C; Kampranis, SC; Kottakis, F; Tsichlis, PN
PLoS pathogens
10
e1004136
2014
概要を表示する
Earlier studies had suggested that epigenetic mechanisms play an important role in the control of human cytomegalovirus (HCMV) infection. Here we show that productive HCMV infection is indeed under the control of histone H3K27 trimethylation. The histone H3K27 methyltransferase EZH2, and its regulators JARID2 and NDY1/KDM2B repress GFI1, a transcriptional repressor of the major immediate-early promoter (MIEP) of HCMV. Knocking down EZH2, NDY1/KDM2B or JARID2 relieves the repression and results in the upregulation of GFI1. During infection, the incoming HCMV rapidly downregulates the GFI1 mRNA and protein in both wild-type cells and in cells in which EZH2, NDY1/KDM2B or JARID2 were knocked down. However, since the pre-infection levels of GFI1 in the latter cells are significantly higher, the virus fails to downregulate it to levels permissive for MIEP activation and viral infection. Following the EZH2-NDY1/KDM2B-JARID2-independent downregulation of GFI1 in the early stages of infection, the virus also initiates an EZH2-NDY1/ΚDM2Β-JARID2-dependent program that represses GFI1 throughout the infection cycle. The EZH2 knockdown also delays histone H3K27 trimethylation in the immediate early region of HCMV, which is accompanied by a drop in H3K4 trimethylation that may contribute to the shEZH2-mediated repression of the major immediate early HCMV promoter. These data show that HCMV uses multiple mechanisms to allow the activation of the HCMV MIEP and to prevent cellular mechanisms from blocking the HCMV replication program. | Western Blotting | 24830456
|
Effects of physiological levels of the green tea extract epigallocatechin-3-gallate on breast cancer cells.
Zeng, L; Holly, JM; Perks, CM
Frontiers in endocrinology
5
61
2014
概要を表示する
Physiological concentrations of the green tea extract epigallocatechin-3-gallate (EGCG) caused growth inhibition in estrogen receptor α (ERα)-positive MCF7 cells that was associated with down-regulation of the ERα and reduced insulin-like growth factor binding protein-2 abundance and increased protein abundance of the tumor suppressor genes p53/p21. In contrast to MCF7 cells that have wt p53, EGCG alone did not change cell proliferation or death significantly in another ERα-positive cell line T47D that possesses mutant p53. EGCG increased ERα protein levels and as a consequence, the cells responded significantly better to an ERα antagonist tamoxifen (TAM) in the presence of EGCG. EGCG significantly increased cell death in an ERα-negative cell line, MDA-MB-231 that also possesses mutant p53. EGCG significantly increased the ERα and insulin-like growth factor-I receptor levels and thereby enhanced the sensitivities of the cells to TAM and a blocking antibody targeting the insulin-like growth factor-1 receptor (αIR3). In contrast to MCF7, T47D and MDA-MB-231 breast cancer cells that exhibited significant changes in key molecules involved in breast growth and survival upon treatment with physiological levels of EGCG, the growth, survival, and levels of these proteins in non-malignant breast epithelial cells, MCF10A cells, were not affected. | Western Blotting | 24847310
|
The FGF-1-specific single-chain antibody scFv1C9 effectively inhibits breast cancer tumour growth and metastasis.
Shi, H; Fu, C; Wang, W; Li, Y; Du, S; Cao, R; Chen, J; Sun, D; Zhang, Z; Wang, X; Zhu, X
Journal of cellular and molecular medicine
18
2061-70
2014
概要を表示する
Immunotherapy mediated by recombinant antibodies is an effective therapeutic strategy for a variety of cancers. In a previous study, we demonstrated that the fibroblast growth factor 1 (FGF-1)-specific recombinant antibody scFv1C9 arrests the cell cycle at the G0/G1 transition by blocking the intracrine FGF-1 pathway in breast cancer cells. Here, we further show that the overexpression of scFv1C9 in MCF-7 and MDA-MB-231 breast cancer cells by lentiviral infection resulted in decreased tumourigenicity, tumour growth and lung metastasis through FGF-1 neutralization. We found that scFv1C9 resulted in the up-regulation of p21, which in turn inhibited the expression of CDK2 and blocked cell cycle progression. To explore the potential role of scFv1C9 in vivo, we delivered the gene into solid tumours by electroporation, which resulted in significant inhibition of tumour growth. In tumour tissue sections, immunohistochemical staining of the cellular proliferation marker Ki-67 and the microvessel marker CD31 showed a reduction in the proliferative index and microvessel density, respectively, upon expression of scFv1C9 compared with the appropriate controls. Thus, our data indicate a central role for scFv1C9 in blocking the intracrine pathway of FGF-1, therefore, scFv1C9 could be developed in an effective therapeutic for breast cancer. | Western Blotting | 25124967
|
The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation.
Chuang, MJ; Wu, ST; Tang, SH; Lai, XM; Lai, HC; Hsu, KH; Sun, KH; Sun, GH; Chang, SY; Yu, DS; Hsiao, PW; Huang, SM; Cha, TL
PloS one
8
e73401
2013
概要を表示する
Histone deacetylase inhibitors (HDACIs) have potent anti-cancer activity in a variety of cancer models. Understanding the molecular mechanisms involved in the therapeutic responsiveness of HDACI is needed before its clinical application. This study aimed to determine if a potent HDACI, LBH589 (Panobinostat), had differential therapeutic responsiveness towards LNCaP and PC-3 prostate cancer (PCa) cells. The former showed prometaphase arrest with subsequent apoptosis upon LBH589 treatment, while the latter was less sensitive and had late G2 arrest. The LBH589 treatment down-regulated HDAC6 and sustained ERK activation, and contributed to prometaphase arrest. Mechanistically, LBH589 inhibited HDAC6 activity, caused its dissociation from protein phosphatase PP1α, and increased 14-3-3ζ acetylation. Acetylated 14-3-3ζ released its mask effect on serine 259 of c-Raf and serine 216 of Cdc25C subsequent to de-phosphorylation by PP1α, which contributed to ERK activation. Enhanced ERK activity by LBH589 further down-regulated HDAC6 protein levels and sustained ERK activation by free-forward regulation. The sustained Cdc25C and ERK activation resulted in early M-phase (prometaphase) arrest and subsequent apoptosis in the most sensitive LNCaP cells but not in PC-3 cells. This study provides pre-clinical evidence that HDAC6 may serve as a sensitive therapeutic target in the treatment of prostate cancer with HDACI LBH589 for clinical translation. This study also posits a novel mechanism of HDAC6 participation in regulating the c-Raf-PP1-ERK signaling pathway and contributing to M phase cell-cycle transition. | | 24023871
|
Overexpression of EVI1 interferes with cytokinesis and leads to accumulation of cells with supernumerary centrosomes in G0/1 phase.
Karakaya, K; Herbst, F; Ball, C; Glimm, H; Krämer, A; Löffler, H
Cell cycle (Georgetown, Tex.)
11
3492-503
2012
概要を表示する
Ectopic viral integration site 1 (EVI1), a transcription factor frequently overexpressed in myeloid neoplasias, has been implicated in the generation of malignancy-associated centrosomal aberrations and chromosomal instability. Here, we sought to investigate the underlying cause of centrosome amplification in EVI1-overexpressing cells. We found that overexpression of EVI1-HA in U2OS cells induced supernumerary centrosomes, which were consistently associated with enlarged nuclei or binuclear cells. Live cell imaging experiments identified cytokinesis failure as the underlying cause of this phenotype. In accordance with previous reports, EVI1 overexpression induced a partial cell cycle arrest in G0/1 phase, accompanied by elevated cyclin D1 and p21 levels, reduced Cdk2 activity and activation of the p53 pathway. Supernumerary centrosomes predominantly occurred in resting cells, as identified by low levels of the proliferation marker Ki-67, leading to the conclusion that they result from tetraploidization after cytokinesis failure and are confined to G0/1-arrested tetraploid cells. Depletion of p53 using siRNA revealed that further polyploidization of these cells was inhibited by the p53-dependent tetraploidy checkpoint. | | 22894935
|
Ablation of PRMT6 reveals a role as a negative transcriptional regulator of the p53 tumor suppressor.
Neault, M; Mallette, FA; Vogel, G; Michaud-Levesque, J; Richard, S
Nucleic acids research
40
9513-21
2012
概要を表示する
Arginine methylation of histones is a well-known regulator of gene expression. Protein arginine methyltransferase 6 (PRMT6) has been shown to function as a transcriptional repressor by methylating the histone H3 arginine 2 [H3R2(me2a)] repressive mark; however, few targets are known. To define the physiological role of PRMT6 and to identify its targets, we generated PRMT6(-/-) mouse embryo fibroblasts (MEFs). We observed that early passage PRMT6(-/-) MEFs had growth defects and exhibited the hallmarks of cellular senescence. PRMT6(-/-) MEFs displayed high transcriptional levels of p53 and its targets, p21 and PML. Generation of PRMT6(-/-); p53(-/-) MEFs prevented the premature senescence, suggesting that the induction of senescence is p53-dependent. Using chromatin immunoprecipitation assays, we observed an enrichment of PRMT6 and H3R2(me2a) within the upstream region of Trp53. The PRMT6 association and the H3R2(me2a) mark were lost in PRMT6(-/-) MEFs and an increase in the H3K4(me3) activator mark was observed. Our findings define a new regulator of p53 transcriptional regulation and define a role for PRMT6 and arginine methylation in cellular senescence. | Western Blotting | 22904064
|