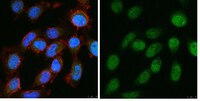
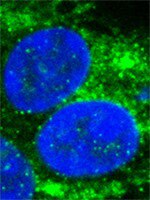
The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma.
Maes, K; De Smedt, E; Lemaire, M; De Raeve, H; Menu, E; Van Valckenborgh, E; McClue, S; Vanderkerken, K; De Bruyne, E
Oncotarget
5
3115-29
2014
概要を表示する
DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi) are under investigation for the treatment of cancer, including the plasma cell malignancy multiple myeloma (MM). Evidence exists that DNA damage and repair contribute to the cytotoxicity mediated by the DNMTi decitabine. Here, we investigated the DNA damage response (DDR) induced by decitabine in MM using 4 human MM cell lines and the murine 5T33MM model. In addition, we explored how the HDACi JNJ-26481585 affects this DDR. Decitabine induced DNA damage (gamma-H2AX foci formation), followed by a G0/G1- or G2/M-phase arrest and caspase-mediated apoptosis. JNJ-26481585 enhanced the anti-MM effect of decitabine both in vitro and in vivo. As JNJ-26481585 did not enhance decitabine-mediated gamma-H2AX foci formation, we investigated the DNA repair response towards decitabine and/or JNJ-26481585. Decitabine augmented RAD51 foci formation (marker for homologous recombination (HR)) and/or 53BP1 foci formation (marker for non-homologous end joining (NHEJ)). Interestingly, JNJ-26481585 negatively affected basal or decitabine-induced RAD51 foci formation. Finally, B02 (RAD51 inhibitor) enhanced decitabine-mediated apoptosis. Together, we report that decitabine-induced DNA damage stimulates HR and/or NHEJ. JNJ-26481585 negatively affects RAD51 foci formation, thereby providing an additional explanation for the combinatory effect between decitabine and JNJ-26481585. | Immunoblotting (Western) | 24833108
 |
Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models.
Ma, CX; Cai, S; Li, S; Ryan, CE; Guo, Z; Schaiff, WT; Lin, L; Hoog, J; Goiffon, RJ; Prat, A; Aft, RL; Ellis, MJ; Piwnica-Worms, H
The Journal of clinical investigation
122
1541-52
2012
概要を表示する
Patients with triple-negative breast cancer (TNBC) - defined by lack of estrogen receptor and progesterone receptor expression as well as lack of human epidermal growth factor receptor 2 (HER2) amplification - have a poor prognosis. There is a need for targeted therapies to treat this condition. TNBCs frequently harbor mutations in TP53, resulting in loss of the G1 checkpoint and reliance on checkpoint kinase 1 (Chk1) to arrest cells in response to DNA damage. Previous studies have shown that inhibition of Chk1 in a p53-deficient background results in apoptosis [corrected] in response to DNA damage. We therefore tested whether inhibition of Chk1 could potentiate the cytotoxicity of the DNA damaging agent irinotecan in TNBC using xenotransplant tumor models. Tumor specimens from patients with TNBC were engrafted into humanized mammary fat pads of immunodeficient mice to create 3 independent human-in-mouse TNBC lines: 1 WT (WU-BC3) and 2 mutant for TP53 (WU-BC4 and WU-BC5). These lines were tested for their response to irinotecan and a Chk1 inhibitor (either UCN-01 or AZD7762), either as single agents or in combination. The combination therapy induced checkpoint bypass and apoptosis in WU-BC4 and WU-BC5, but not WU-BC3, tumors. Moreover, combination therapy inhibited tumor growth and prolonged survival of mice bearing the WU-BC4 line, but not the WU-BC3 line. In addition, knockdown of p53 sensitized WU-BC3 tumors to the combination therapy. These results demonstrate that p53 is a major determinant of how TNBCs respond to therapies that combine DNA damage with Chk1 inhibition. | Western Blotting | 22446188
|