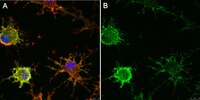
Dynamic expression of srGAP2 in cell nuclei and cytoplasm during the differentiation of rat neural stem cells in vitro
Qian Jiao 1 , Li Wang 1 , Zhichao Zhang 1 , Yuanyuan Wang 1 , Hanqi Yan 1 , Wen Ma 1 , Weilin Jin 2 , Haixia Lu 1 , Yong Liu
Mol Med Rep
14(5)
4599-4605
2016
概要を表示する
Different SLIT-ROBO Rho GTPase-activating proteins (srGAPs) have different levels of expression and diverse functions during neural development. Although srGAP2 is expressed in developmental brain tissue, little is known about its influence on cellular development of the nervous system. In the current study, dynamic expression of endogenous srGAP2 during neural stem cell/progenitor cell (NSC/NPC) differentiation in vitro was investigated in order to elucidate the association between the dynamic expression of srGAP2 and neural development. srGAP2 was expressed in undifferentiated NSCs/NPCs, and differentiated neurons and astrocytes with distinct expression patterns. In conjunction with the differentiation of NSCs/NPCs in vitro, the number of srGAP2+ cells markedly reduced. The percentage of srGAP2+ cells in the population of nestin+ and β‑tubulin III+ cells was significantly downregulated while in the population of glial fibrillary acidic protein‑positive cells, almost all cells were srGAP2+. srGAP2 was predominantly expressed in the cell nucleus in all cell types. srGAP2 was also weakly expressed in the cytoplasm of nestin+ and β‑tubulin III+ cells at 3 and 7 days in vitro. However levels were gradually downregulated during the process of differentiation and almost disappeared in β‑tubulin III+ cells at 14 days. The results from the present study suggest that srGAP2 is involved in regulating NSC/NPC differentiation during neural development. The translocation of srGAP2 in the cytoplasm and cell nucleus in different cell types may function as a director in decisions regarding cell fate. | 27748913
 |
Overexpression of heparanase lowers the amyloid burden in amyloid-β precursor protein transgenic mice
Charlotte B Jendresen 1 , Hao Cui 2 , Xiao Zhang 3 , Israel Vlodavsky 4 , Lars N G Nilsson 5 , Jin-Ping Li 6
J Biol Chem
290(8)
5053-64
2015
概要を表示する
Heparan sulfate (HS) and HS proteoglycans (HSPGs) colocalize with amyloid-β (Aβ) deposits in Alzheimer disease brain and in Aβ precursor protein (AβPP) transgenic mouse models. Heparanase is an endoglycosidase that specifically degrades the unbranched glycosaminoglycan side chains of HSPGs. The aim of this study was to test the hypothesis that HS and HSPGs are active participators of Aβ pathogenesis in vivo. We therefore generated a double-transgenic mouse model overexpressing both human heparanase and human AβPP harboring the Swedish mutation (tgHpa*Swe). Overexpression of heparanase did not affect AβPP processing because the steady-state levels of Aβ1-40, Aβ1-42, and soluble AβPP β were the same in 2- to 3-month-old double-transgenic tgHpa*Swe and single-transgenic tgSwe mice. In contrast, the Congo red-positive amyloid burden was significantly lower in 15-month-old tgHpa*Swe brain than in tgSwe brain. Likewise, the Aβ burden, measured by Aβx-40 and Aβx-42 immunohistochemistry, was reduced significantly in tgHpa*Swe brain. The intensity of HS-stained plaques correlated with the Aβx-42 burden and was reduced in tgHpa*Swe mice. Moreover, the HS-like molecule heparin facilitated Aβ1-42-aggregation in an in vitro Thioflavin T assay. The findings suggest that HSPGs contribute to amyloid deposition in tgSwe mice by increasing Aβ fibril formation because heparanase-induced fragmentation of HS led to a reduced amyloid burden. Therefore, drugs interfering with Aβ-HSPG interactions might be a potential strategy for Alzheimer disease treatment. | 25548284
|
The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene
Marta Luna-Sánchez 1 , Elena Díaz-Casado 1 , Emanuele Barca 2 , Miguel Ángel Tejada 3 , Ángeles Montilla-García 3 , Enrique Javier Cobos 3 , Germaine Escames 1 , Dario Acuña-Castroviejo 1 , Catarina M Quinzii 2 , Luis Carlos López
EMBO Mol Med
7(5)
670-87
2015
概要を表示する
Primary coenzyme Q10 (CoQ10) deficiency is due to mutations in genes involved in CoQ biosynthesis. The disease has been associated with five major phenotypes, but a genotype-phenotype correlation is unclear. Here, we compare two mouse models with a genetic modification in Coq9 gene (Coq9(Q95X) and Coq9(R239X)), and their responses to 2,4-dihydroxybenzoic acid (2,4-diHB). Coq9(R239X) mice manifest severe widespread CoQ deficiency associated with fatal encephalomyopathy and respond to 2,4-diHB increasing CoQ levels. In contrast, Coq9(Q95X) mice exhibit mild CoQ deficiency manifesting with reduction in CI+III activity and mitochondrial respiration in skeletal muscle, and late-onset mild mitochondrial myopathy, which does not respond to 2,4-diHB. We show that these differences are due to the levels of COQ biosynthetic proteins, suggesting that the presence of a truncated version of COQ9 protein in Coq9(R239X) mice destabilizes the CoQ multiprotein complex. Our study points out the importance of the multiprotein complex for CoQ biosynthesis in mammals, which may provide new insights to understand the genotype-phenotype heterogeneity associated with human CoQ deficiency and may have a potential impact on the treatment of this mitochondrial disorder. | 25802402
|
Apoptosis of neurons and oligodendrocytes in the spinal cord of spinal hyperostotic mouse (twy/twy): possible pathomechanism of human cervical compressive myelopathy
Kenzo Uchida 1 , Hideaki Nakajima, Shuji Watanabe, Takafumi Yayama, Alexander Rodriguez Guerrero, Tomoo Inukai, Takayuki Hirai, Daisuke Sugita, William E Johnson, Hisatoshi Baba
Eur Spine J
21(3)
490-7
2012
概要を表示する
Introduction: Cervical compressive myelopathy is the most serious complication of cervical spondylosis or ossification of the posterior longitudinal ligament (OPLL) and the most frequent cause of spinal cord dysfunction. There is little information on the exact pathophysiological mechanism responsible for the progressive loss of neural tissue in the spinal cord of such patients. In this study, we used the spinal hyperostotic mouse (twy/twy) as a suitable model of human spondylosis, and OPLL to investigate the cellular and molecular changes in the spinal cord. Mutant twy/twy mouse developed ossification of the ligamentum flavum at C2-C3 and exhibited progressive paralysis. <br />Materials and methods: The mutant twy/twy mice, aged 16 and 24 weeks, were used in the present study. The cervical spinal cord was analyzed histologically and immunohistochemically. <br />Results: We observed that a significant correlation between the proportion of apoptotic oligodendrocytes in the compressed area of the spinal cord and the magnitude of cord compression. Immunohistochemical analysis indicated overexpression of TNFR1, CD95, and p75NTR in the twy/twy mice, which was localized by the immunofluorescence in the neurons and oligodendrocytes. <br />Conclusion: The expression of such factors seems to play at least some role in the apoptotic process, which probably contributes to axonal degeneration and demyelination in the twy/twy mice spinal cords with severe compression. | 21935678
|