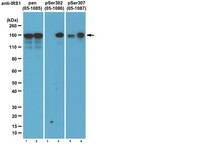
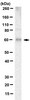
PTEN is a protein tyrosine phosphatase for IRS1.
Shi, Y; Wang, J; Chandarlapaty, S; Cross, J; Thompson, C; Rosen, N; Jiang, X
Nature structural & molecular biology
21
522-7
2014
概要を表示する
The biological function of the PTEN tumor suppressor is mainly attributed to its lipid phosphatase activity. This study demonstrates that mammalian PTEN is a protein tyrosine phosphatase that selectively dephosphorylates insulin receptor substrate-1 (IRS1), a mediator of insulin and IGF signals. IGF signaling was defective in cells lacking NEDD4, a PTEN ubiquitin ligase, whereas AKT activation triggered by EGF or serum was unimpaired. Defective IGF signaling caused by NEDD4 deletion, including phosphorylation of IRS1 and AKT, was rescued by PTEN ablation. We demonstrate the nature of PTEN as an IRS1 phosphatase by direct biochemical analysis and cellular reconstitution, showing that NEDD4 supports insulin-mediated glucose metabolism and is required for the proliferation of IGF1 receptor-dependent but not EGF receptor-dependent tumor cells. Thus, PTEN is a protein phosphatase for IRS1, and its antagonism by NEDD4 promotes signaling by IGF and insulin. | 24814346
 |
Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation.
Hançer, NJ; Qiu, W; Cherella, C; Li, Y; Copps, KD; White, MF
The Journal of biological chemistry
289
12467-84
2014
概要を表示する
IRS1 and IRS2 are key substrates of the insulin receptor tyrosine kinase. Mass spectrometry reveals more than 50 phosphorylated IRS1 serine and threonine residues (Ser(P)/Thr(P) residues) in IRS1 from insulin-stimulated cells or human tissues. We investigated a subset of IRS1 Ser(P)/Thr(P) residues using a newly developed panel of 25 phospho-specific monoclonal antibodies (αpS/TmAb(Irs1)). CHO cells overexpressing the human insulin receptor and rat IRS1 were stimulated with insulin in the absence or presence of inhibitors of the PI3K → Akt → mechanistic target of rapamycin (mTOR) → S6 kinase or MEK pathways. Nearly all IRS1 Ser(P)/Thr(P) residues were stimulated by insulin and significantly suppressed by PI3K inhibition; fewer were suppressed by Akt or mTOR inhibition, and none were suppressed by MEK inhibition. Insulin-stimulated Irs1 tyrosine phosphorylation (Tyr(P)(Irs1)) was enhanced by inhibition of the PI3K → Akt → mTOR pathway and correlated with decreased Ser(P)-302(Irs1), Ser(P)-307(Irs1), Ser(P)-318(Irs1), Ser(P)-325(Irs1), and Ser(P)-346(Irs1). Metabolic stress modeled by anisomycin, thapsigargin, or tunicamycin increased many of the same Ser(P)/Thr(P) residues as insulin, some of which (Ser(P)-302(Irs1), Ser(P)-307(Irs1), and four others) correlated significantly with impaired insulin-stimulated Tyr(P)(Irs1). Thus, IRS1 Ser(P)/Thr(P) is an integrated response to insulin stimulation and metabolic stress, which associates with reduced Tyr(P)(Irs1) in CHO(IR)/IRS1 cells. | 24652289
|
Up-regulated miR-145 expression inhibits porcine preadipocytes differentiation by targeting IRS1.
Guo, Y; Chen, Y; Zhang, Y; Zhang, Y; Chen, L; Mo, D
International journal of biological sciences
8
1408-17
2012
概要を表示する
Generally, most miRNAs that were up-regulated during differentiation promoted adipogenesis, but our research indicated that up-regulation of miR-145 in porcine preadipocytes did not promote but inhibit adipogenesis. In this study, miR-145 was significantly up-regulated during porcine dedifferentiated fat (DFAT) cells differentiation. In miR-145 overexpressed DFAT cells, adipogenesis was inhibited and triglycerides accumulation was decreased after hormone stimulation (Pless than 0.05). Furthermore, up-regulation of miR-145 expression repressed induction of mRNA levels of adipogenic markers, such as CCAAT/enhancer-binding protein α (C/EBPα), and peroxisome proliferator-activated receptor γ2 (PPARγ2). These effects caused by miR-145 overexpression were mediated by Insulin receptor substrate 1 (IRS1) as a mechanism. These data suggested that induced miR-145 expression during differentiation could inhibit adipogenesis by targeting IRS1, and miR-145 may be novel agent for adipose tissue engineering. | 23197937
|
Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2.
Ye, L; Varamini, B; Lamming, DW; Sabatini, DM; Baur, JA
Frontiers in genetics
3
177
2012
概要を表示する
Rapamycin, an inhibitor of mTOR complex 1 (mTORC1), improves insulin sensitivity in acute studies in vitro and in vivo by disrupting a negative feedback loop mediated by S6 kinase. We find that rapamycin has a clear biphasic effect on insulin sensitivity in C2C12 myotubes, with enhanced responsiveness during the first hour that declines to almost complete insulin resistance by 24-48 h. We and others have recently observed that chronic rapamycin treatment induces insulin resistance in rodents, at least in part due to disruption of mTORC2, an mTOR-containing complex that is not acutely sensitive to the drug. Chronic rapamycin treatment may also impair insulin action via the inhibition of mTORC1-dependent mitochondrial biogenesis and activity, which could result in a buildup of lipid intermediates that are known to trigger insulin resistance. We confirmed that rapamycin inhibits expression of PGC-1α, a key mitochondrial transcription factor, and acutely reduces respiration rate in myotubes. However, rapamycin did not stimulate phosphorylation of PKCθ, a central mediator of lipid-induced insulin resistance. Instead, we found dramatic disruption of mTORC2, which coincided with the onset of insulin resistance. Selective inhibition of mTORC1 or mTORC2 by shRNA-mediated knockdown of specific components (Raptor and Rictor, respectively) confirmed that mitochondrial effects of rapamycin are mTORC1-dependent, whereas insulin resistance was recapitulated only by knockdown of mTORC2. Thus, mTORC2 disruption, rather than inhibition of mitochondria, causes insulin resistance in rapamycin-treated myotubes, and this system may serve as a useful model to understand the effects of rapamycin on mTOR signaling in vivo. | 22973301
|