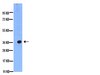
RE-IIBP Methylates H3K79 and Induces MEIS1-mediated Apoptosis via H2BK120 Ubiquitination by RNF20.
Woo Park, J; Kim, KB; Kim, JY; Chae, YC; Jeong, OS; Seo, SB
Scientific reports
5
12485
2015
Mostra il sommario
Histone lysine methylation contributes to transcriptional regulation by serving as a platform for the recruitment of various cofactors. Intense studies have been conducted for elucidating the functional meaning of H3K79 methylation, and to date, the only known HMTase responsible for the modification was DOT1L. In this study, we report that the MMSET isoform RE-IIBP has HMTase activity for H3K79. It was uncovered that RE-IIBP up-regulates MEIS1 transcription through H3K79 methylation via recruitment to the MEIS1 promoter. By means of proteomic and biochemical analysis, association of RE-IIBP with the E3 ubiquitin ligase RNF20 was demonstrated for synergistic activation of MEIS1 transcription via H3K79 HMTase activity. Furthermore, It was observed that RE-IIBP induces MEIS1-mediated apoptosis, which was dependent on H2BK120 ubiquitination by RNF20. These findings suggest RE-IIBP as another candidate for further studies to elucidate the mechanism of H3K79 methylation and its biological functions. | | | 26206755
 |
Epigenetic regulation of traf2- and Nck-interacting kinase (TNIK) in polycystic ovary syndrome.
Li, D; Jiao, J; Zhou, YM; Wang, XX
American journal of translational research
7
1152-60
2015
Mostra il sommario
Emerging evidence has led to considerable interest in the role of Traf2- and Nck-interacting kinase (TNIK) in polycystic ovary syndrome (PCOS) development. However, the epigenetic mechanism regulating TNIK transcription remains largely unknown. Here, we show that (i) TNIK mRNA expression is significantly increased in PCOS ovarian tissues, compared to normal ovarian tissues; (ii) PCOS ovarian tissues exhibit a hypermethylation pattern at the cg10180092 site, (iii) and cg10180092 is the critical site for the transcriptional regulation of TNIK. Mechanistically, hypermethylated cg10180092 site-mediated loss of holocarboxylase synthetase (HLCS)-related H3K9me enrichment activated TNIK transcription in PCOS ovarian tissues. Notably, a substantial body of evidence indicates that DNA hypermethylation is an alternative mechanism for gene inactivation, and a new role for DNA hypermethylationmediated TNIK activating was observed in this study. This may improve our understanding of divergent transcriptional regulation in the initiation and progression of TNIK-related PCOS. | | | 26279758
|
Chromatin organization and cytological features of carnivorous Genlisea species with large genome size differences.
Tran, TD; Cao, HX; Jovtchev, G; Novák, P; Vu, GT; Macas, J; Schubert, I; Fuchs, J
Frontiers in plant science
6
613
2015
Mostra il sommario
The monophyletic carnivorous genus Genlisea (Lentibulariaceae) is characterized by a bi-directional genome size evolution resulting in a 25-fold difference in nuclear DNA content. This is one of the largest ranges found within a genus so far and makes Genlisea an interesting subject to study mechanisms of genome and karyotype evolution. Genlisea nigrocaulis, with 86 Mbp one of the smallest plant genomes, and the 18-fold larger genome of G. hispidula (1,550 Mbp) possess identical chromosome numbers (2n = 40) but differ considerably in chromatin organization, nuclear and cell size. Interphase nuclei of G. nigrocaulis and of related species with small genomes, G. aurea (133 Mbp, 2n ≈ 104) and G. pygmaea (179 Mbp, 2n = 80), are hallmarked by intensely DAPI-stained chromocenters, carrying typical heterochromatin-associated methylation marks (5-methylcytosine, H3K9me2), while in G. hispidula and surprisingly also in the small genome of G. margaretae (184 Mbp, 2n = 38) the heterochromatin marks are more evenly distributed. Probes of tandem repetitive sequences together with rDNA allow the unequivocal discrimination of 13 out of 20 chromosome pairs of G. hispidula. One of the repetitive sequences labeled half of the chromosome set almost homogenously supporting an allopolyploid status of G. hispidula and its close relative G. subglabra (1,622 Mbp, 2n = 40). In G. nigrocaulis 11 chromosome pairs could be individualized using a combination of rDNA and unique genomic probes. The presented data provide a basis for future studies of karyotype evolution within the genus Genlisea. | | | 26347752
|
Sgf73, a subunit of SAGA complex, is required for the assembly of RITS complex in fission yeast.
Deng, X; Zhou, H; Zhang, G; Wang, W; Mao, L; Zhou, X; Yu, Y; Lu, H
Scientific reports
5
14707
2015
Mostra il sommario
RNA interference (RNAi) is a widespread gene-silencing mechanism and is required for heterochromatin assembly in a variety of organisms. The RNA-induced transcriptional silencing complex (RITS), composed of Ago1, Tas3 and Chp1, is a key component of RNAi machinery in fission yeast that connects short interference RNA (siRNA) and heterochromatin formation. However, the process by which RITS is assembled is not well understood. Here, we identified Sgf73, a subunit of the SAGA co-transcriptional complex, is required for pericentromeric heterochromatin silencing and the generation of siRNA. This novel role of Sgf73 is independent of enzymatic activities or structural integrity of SAGA. Instead, Sgf73 is physically associated with Ago1 and Chp1. The interactions among the subunits of the RITS, including those between Tas3 and Chp1, between Chp1 and Ago1, between Ago1 and Tas3, were all impaired by the deletion of sgf73(+). Consistently, the recruitment of Ago1 and Chp1 to the pericentromeric region was abolished in sgf73Δ cells. Our study unveils a moonlighting function of a SAGA subunit. It suggests Sgf73 is a novel factor that promotes assembly of RITS and RNAi-mediated heterochromatin formation. | | | 26443059
|
The histone H3K9 demethylase Kdm3b is required for somatic growth and female reproductive function.
Liu, Z; Chen, X; Zhou, S; Liao, L; Jiang, R; Xu, J
International journal of biological sciences
11
494-507
2015
Mostra il sommario
Kdm3b is a Jumonji C domain-containing protein that demethylates mono- and di-methylated lysine 9 of histone H3 (H3K9me1 and H3K9me2). Although the enzyme activity of Kdm3b is well characterized in vitro, its genetic and physiological function remains unknown. Herein, we generated Kdm3b knockout (Kdm3bKO) mice and observed restricted postnatal growth and female infertility in these mice. We found that Kdm3b ablation decreased IGFBP-3 expressed in the kidney by 53% and significantly reduced IGFBP-3 in the blood, which caused an accelerated degradation of IGF-1 and a 36% decrease in circulating IGF-1 concentration. We also found Kdm3b was highly expressed in the female reproductive organs including ovary, oviduct and uterus. Knockout of Kdm3b in female mice caused irregular estrous cycles, decreased 45% of the ovulation capability and 47% of the fertilization rate, and reduced 44% of the uterine decidual response, which were accompanied with a more than 50% decrease in the circulating levels of the 17beta-estradiol. Importantly, these female reproductive phenotypes were associated with significantly increased levels of H3K9me1/2/3 in the ovary and uterus. These results demonstrate that Kdm3b-mediated H3K9 demethylation plays essential roles in maintenance of the circulating IGF-1, postnatal somatic growth, circulating 17beta-estradiol, and female reproductive function. | | | 25892958
|
H3K9 methyltransferase G9a negatively regulates UHRF1 transcription during leukemia cell differentiation.
Kim, KB; Son, HJ; Choi, S; Hahm, JY; Jung, H; Baek, HJ; Kook, H; Hahn, Y; Kook, H; Seo, SB
Nucleic acids research
43
3509-23
2015
Mostra il sommario
Histone H3K9 methyltransferase (HMTase) G9a-mediated transcriptional repression is a major epigenetic silencing mechanism. UHRF1 (ubiquitin-like with PHD and ring finger domains 1) binds to hemimethylated DNA and plays an essential role in the maintenance of DNA methylation. Here, we provide evidence that UHRF1 is transcriptionally downregulated by H3K9 HMTase G9a. We found that increased expression of G9a along with transcription factor YY1 specifically represses UHRF1 transcription during TPA-mediated leukemia cell differentiation. Using ChIP analysis, we found that UHRF1 was among the transcriptionally silenced genes during leukemia cell differentiation. Using a DNA methylation profiling array, we discovered that the UHRF1 promoter was hypomethylated in samples from leukemia patients, further supporting its overexpression and oncogenic activity. Finally, we showed that G9a regulates UHRF1-mediated H3K23 ubiquitination and proper DNA replication maintenance. Therefore, we propose that H3K9 HMTase G9a is a specific epigenetic regulator of UHRF1. | | | 25765655
|
Alternative splicing regulates the expression of G9A and SUV39H2 methyltransferases, and dramatically changes SUV39H2 functions.
Mauger, O; Klinck, R; Chabot, B; Muchardt, C; Allemand, E; Batsché, E
Nucleic acids research
43
1869-82
2015
Mostra il sommario
Alternative splicing is the main source of proteome diversity. Here, we have investigated how alternative splicing affects the function of two human histone methyltransferases (HMTase): G9A and SUV39H2. We show that exon 10 in G9A and exon 3 in SUV39H2 are alternatively included in a variety of tissues and cell lines, as well as in a different species. The production of these variants is likely tightly regulated because both constitutive and alternative splicing factors control their splicing profiles. Based on this evidence, we have assessed the link between the inclusion of these exons and the activity of both enzymes. We document that these HMTase genes yield several protein isoforms, which are likely issued from alternative splicing regulation. We demonstrate that inclusion of SUV39H2 exon 3 is a determinant of the stability, the sub-nuclear localization, and the HMTase activity. Genome-wide expression analysis further revealed that alternative inclusion of SUV39H2 exon 3 differentially modulates the expression of target genes. Our data also suggest that a variant of G9A may display a function that is independent of H3K9 methylation. Our work emphasizes that expression and function of genes are not collinear; therefore alternative splicing must be taken into account in any functional study. | Western Blotting | | 25605796
|
The demethylase JMJD2C localizes to H3K4me3-positive transcription start sites and is dispensable for embryonic development.
Pedersen, MT; Agger, K; Laugesen, A; Johansen, JV; Cloos, PA; Christensen, J; Helin, K
Molecular and cellular biology
34
1031-45
2014
Mostra il sommario
The histone demethylase JMJD2C, also known as KDM4C/GASC1, has activity against methylated H3K9 and H3K36 and is amplified and/or overexpressed in human cancers. By the generation of Jmjd2c knockout mice, we demonstrate that loss of Jmjd2c is compatible with cellular proliferation, embryonic stem cell (ESC) self-renewal, and embryonic development. Moreover, we report that JMJD2C localizes to H3K4me3-positive transcription start sites in both primary cells and in the human carcinoma KYSE150 cell line containing an amplification of the JMJD2C locus. Binding is dependent on the double Tudor domain of JMJD2C, which recognizes H3K4me3 but not H4K20me2/me3 in vitro, showing a binding specificity different from that of the double Tudor domains of JMJD2A and JMJD2B. Depletion of JMJD2C in KYSE150 cells has a modest effect on H3K9me3 and H3K36me3 levels but impairs proliferation and leads to deregulated expression of a subset of target genes involved in cell cycle progression. Taking these findings together, we show that JMJD2C is targeted to H3K4me3-positive transcription start sites, where it can contribute to transcriptional regulation, and report that the putative oncogene JMJD2C generally is not required for cellular proliferation or embryonic development. | | | 24396064
|
Comparative analysis of genome-wide chromosomal histone modification patterns in maize cultivars and their wild relatives.
He, S; Yan, S; Wang, P; Zhu, W; Wang, X; Shen, Y; Shao, K; Xin, H; Li, S; Li, L
PloS one
9
e97364
2014
Mostra il sommario
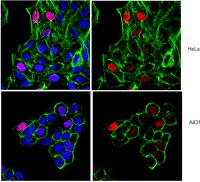
Recent advances demonstrate that epigenome changes can also cause phenotypic diversity and can be heritable across generations, indicating that they may play an important role in evolutionary processes. In this study, we analyzed the chromosomal distribution of several histone modifications in five elite maize cultivars (B73, Mo17, Chang7-2, Zheng58, ZD958) and their two wild relatives (Zea mays L. ssp. parviglumis and Zea nicaraguensis) using a three-dimensional (3D) epigenome karyotyping approach by combining immunostaining and 3D reconstruction with deconvolution techniques. The distribution of these histone modifications along chromosomes demonstrated that the histone modification patterns are conserved at the chromosomal level and have not changed significantly following domestication. The comparison of histone modification patterns between metaphase chromosomes and interphase nuclei showed that some of the histone modifications were retained as the cell progressed from interphase into metaphase, although remodelling existed. This study will increase comprehension of the function of epigenetic modifications in the structure and evolution of the maize genome. | Immunocytochemistry | | 24819606
|
Histone H3S10 phosphorylation by the JIL-1 kinase in pericentric heterochromatin and on the fourth chromosome creates a composite H3S10phK9me2 epigenetic mark.
Wang, Chao, et al.
Chromosoma, (2014)
2014
Mostra il sommario
The JIL-1 kinase mainly localizes to euchromatic interband regions of polytene chromosomes and is the kinase responsible for histone H3S10 phosphorylation at interphase in Drosophila. However, recent findings raised the possibility that the binding of some H3S10ph antibodies may be occluded by the H3K9me2 mark obscuring some H3S10 phosphorylation sites. Therefore, we have characterized an antibody to the epigenetic H3S10phK9me2 double mark as well as three commercially available H3S10ph antibodies. The results showed that for some H3S10ph antibodies their labeling indeed can be occluded by the concomitant presence of the H3K9me2 mark. Furthermore, we demonstrate that the double H3S10phK9me2 mark is present in pericentric heterochromatin as well as on the fourth chromosome of wild-type polytene chromosomes but not in preparations from JIL-1 or Su(var)3-9 null larvae. Su(var)3-9 is a methyltransferase mediating H3K9 dimethylation. Furthermore, the H3S10phK9me2 labeling overlapped with that of the non-occluded H3S10ph antibodies as well as with H3K9me2 antibody labeling. Interestingly, when a Lac-I-Su(var)3-9 transgene is overexpressed, it upregulates H3K9me2 dimethylation on the chromosome arms creating extensive ectopic H3S10phK9me2 marks suggesting that the H3K9 dimethylation occurred at euchromatic H3S10ph sites. This is further supported by the finding that under these conditions euchromatic H3S10ph labeling by the occluded antibodies was abolished. Thus, our findings indicate a novel role for the JIL-1 kinase in epigenetic regulation of heterochromatin in the context of the chromocenter and fourth chromosome by creating a composite H3S10phK9me2 mark together with the Su(var)3-9 methyltransferase. | | | 24429699
|