Identification and characterization of mouse otic sensory lineage genes.
Hartman, BH; Durruthy-Durruthy, R; Laske, RD; Losorelli, S; Heller, S
Frontiers in cellular neuroscience
9
79
2015
Mostra il sommario
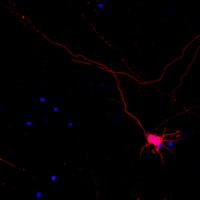
Vertebrate embryogenesis gives rise to all cell types of an organism through the development of many unique lineages derived from the three primordial germ layers. The otic sensory lineage arises from the otic vesicle, a structure formed through invagination of placodal non-neural ectoderm. This developmental lineage possesses unique differentiation potential, giving rise to otic sensory cell populations including hair cells, supporting cells, and ganglion neurons of the auditory and vestibular organs. Here we present a systematic approach to identify transcriptional features that distinguish the otic sensory lineage (from early otic progenitors to otic sensory populations) from other major lineages of vertebrate development. We used a microarray approach to analyze otic sensory lineage populations including microdissected otic vesicles (embryonic day 10.5) as well as isolated neonatal cochlear hair cells and supporting cells at postnatal day 3. Non-otic tissue samples including periotic tissues and whole embryos with otic regions removed were used as reference populations to evaluate otic specificity. Otic populations shared transcriptome-wide correlations in expression profiles that distinguish members of this lineage from non-otic populations. We further analyzed the microarray data using comparative and dimension reduction methods to identify individual genes that are specifically expressed in the otic sensory lineage. This analysis identified and ranked top otic sensory lineage-specific transcripts including Fbxo2, Col9a2, and Oc90, and additional novel otic lineage markers. To validate these results we performed expression analysis on select genes using immunohistochemistry and in situ hybridization. Fbxo2 showed the most striking pattern of specificity to the otic sensory lineage, including robust expression in the early otic vesicle and sustained expression in prosensory progenitors and auditory and vestibular hair cells and supporting cells. | Immunohistochemistry | 25852475
 |
Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner.
Lim, KS; Lim, KJ; Price, AC; Orr, BA; Eberhart, CG; Bar, EE
Oncogene
33
4433-41
2014
Mostra il sommario
Hypoxic regions are frequent in glioblastoma (GBM), the most common type of malignant adult brain tumor, and increased levels of tumor hypoxia have been associated with worse clinical outcomes. To unmask genes important in hypoxia, we treated GBM neurospheres in hypoxia and identified monocarboxylate transporter-4 (MCT4) as one of the most upregulated genes. To investigate the clinical importance of MCT4 in GBM, we examined clinical outcomes and found that MCT4 overexpression is associated with shorter patient survival. Consistent with this, MCT4 upregulation correlated with the aggressive mesenchymal subset of GBM, and MCT4 downregulation correlated with the less aggressive G-CIMP (Glioma CpG Methylator Phenotype) subset of GBM. Immunohistochemical analysis of tissue microarrays confirmed that MCT4 protein levels were increased in high-grade as compared with lower-grade astrocytomas, further suggesting that MCT4 is a clinically relevant target. To test the requirement for MCT4 in vitro, we transduced neurospheres with lentiviruses encoding short-hairpin RNAs (shRNAs) against MCT4, resulting in growth inhibition of 50-80% under hypoxia in two lines. MCT4 knockdown was associated with a decreased percentage of cells expressing the stem-cell marker CD133 and increased apoptotic fraction. We also found that flow-sorted CD133-positive cells had almost sixfold higher MCT4 levels than CD133-negative cells, suggesting that the stem-like population might have a greater requirement for MCT4. Most importantly, MCT4 silencing also slowed GBM intracranial xenograft growth in vivo. Interestingly, whereas MCT4 is a well-characterized lactate exporter, we found that both intracellular and extracellular lactate levels did not change following MCT4 silencing, suggesting a novel lactate export-independent mechanism for growth inhibition in GBMs. To identify this potential mechanism, we performed microarray analysis on control and shMCT4-expressing neurospheres and found a dramatic reduction in the expression of multiple Hypoxia-Inducible Factor (HIF)-regulated genes following MCT4 knockdown. The overall reduction in HIF transcriptional response was further validated using a hypoxia response element (HRE)-dependent green-fluorescent protein (GFP) reporter line. | | 24077291
|
Enhancement of brain-type creatine kinase activity ameliorates neuronal deficits in Huntington's disease.
Lin, YS; Cheng, TH; Chang, CP; Chen, HM; Chern, Y
Biochimica et biophysica acta
1832
742-53
2013
Mostra il sommario
Huntington's disease (HD) is a hereditary neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin (HTT) gene. Brain-type creatine kinase (CKB) is an enzyme involved in energy homeostasis via the phosphocreatine-creatine kinase system. Although downregulation of CKB was previously reported in brains of HD mouse models and patients, such regulation and its functional consequence in HD are not fully understood. In the present study, we demonstrated that levels of CKB found in both the soma and processes were markedly reduced in primary neurons and brains of HD mice. We show for the first time that mutant HTT (mHTT) suppressed the activity of the promoter of the CKB gene, which contributes to the lowered CKB expression in HD. Exogenous expression of wild-type CKB, but not a dominant negative CKB mutant, rescued the ATP depletion, aggregate formation, impaired proteasome activity, and shortened neurites induced by mHTT. These findings suggest that negative regulation of CKB by mHTT is a key event in the pathogenesis of HD and contributes to the neuronal dysfunction associated with HD. In addition, besides dietary supplementation with the CKB substrate, strategies aimed at increasing CKB expression might lead to the development of therapeutic treatments for HD. | | 23416527
|
Expression and subcellular distribution of imp13 are regulated in brain development.
You, P; Peng, Z; Wang, Y; Tao, T
In vitro cellular & developmental biology. Animal
49
346-53
2013
Mostra il sommario
Imp13, a member of importin-β superfamily, is found to be one of two bidirectional transport receptors in many nuclear transport activities in mammals. Several cargoes of imp13 have been identified; most of these are essential factors involved in cell cycle and development. The expression and localization of imp13 may influence its cargoes in playing their roles in appropriate time and space. To gain insight into the role of imp13 in brain development, we generated an anti-imp13 polyclonal antibody and investigated the expressions of imp13 in mouse embryonic brains during development, including E13.5, E15.5, E17.5, P0, and adult, at both transcriptional and translational levels. In addition, we performed immunohistochemical analysis and revealed that imp13 tends to be localized in the cytoplasm at the early stages and relocates into the nucleus at the late stages in neuronal cells of mouse brains. These findings suggested that the expression and localization of imp13 in brain tissues are regulated developmentally, which extends our knowledge of the dynamic presence of imp13. These observations also imply that imp13 contributes to the neural cell-specific cargo trafficking and potentially to other functions during brain development. | | 23605716
|
L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the 'collusion' hypothesis for increased extracellular L-glutamate concentration in neuroinflammation.
Takaki, J; Fujimori, K; Miura, M; Suzuki, T; Sekino, Y; Sato, K
Journal of neuroinflammation
9
275
2011
Mostra il sommario
In the central nervous system, astrocytic L-glutamate (L-Glu) transporters maintain extracellular L-Glu below neurotoxic levels, but their function is impaired with neuroinflammation. Microglia become activated with inflammation; however, the correlation between activated microglia and the impairment of L-Glu transporters is unknown.We used a mixed culture composed of astrocytes, microglia, and neurons. To quantify L-Glu transporter function, we measured the extracellular L-Glu that remained 30 min after an application of L-Glu to the medium (the starting concentration was 100 μM). We determined the optimal conditions of lipopolysaccharide (LPS) treatment to establish an inflammation model without cell death. We examined the predominant subtypes of L-Glu transporters and the changes in the expression levels of these transporters in this inflammation model. We then investigated the role of activated microglia in the changes in L-Glu transporter expression and the underlying mechanisms in this inflammation model.Because LPS (10 ng/mL, 72 h) caused a significant increase in the levels of L-Glu remaining but did not affect cell viability, we adopted this condition for our inflammation model without cell death. GLAST was the predominant L-Glu transporter subtype, and its expression decreased in this inflammation model. As a result of their release of L-Glu, activated microglia were shown to be essential for the significant decrease in L-Glu uptake. The serial application of L-Glu caused a significant decrease in L-Glu uptake and GLAST expression in the astrocyte culture. The hemichannel inhibitor carbenoxolone (CBX) inhibited L-Glu release from activated microglia and ameliorated the decrease in GLAST expression in the inflammation model. In addition, the elevation of the astrocytic intracellular L-Glu itself caused the downregulation of GLAST.Our findings suggest that activated microglia trigger the elevation of extracellular L-Glu through their own release of L-Glu, and astrocyte L-Glu transporters are downregulated as a result of the elevation of astrocytic intracellular L-Glu levels, causing a further increase of extracellular L-Glu. Our data suggest the new hypothesis that activated microglia collude with astrocytes to cause the elevation of extracellular L-Glu in the early stages of neuroinflammation. | | 23259598
|
The temporal and spatial expression pattern of the LGI1 epilepsy predisposition gene during mouse embryonic cranial development.
Silva, J; Wang, G; Cowell, JK
BMC neuroscience
12
43
2010
Mostra il sommario
Mutations in the LGI1 gene predispose to a rare, hereditary form of temporal epilepsy. Currently, little is known about the temporal and spatial expression pattern of Lgi1 during normal embryogenesis and so to define this more clearly we used a transgenic mouse line that expresses GFP under the control of Lgi1 cis-regulatory elements.During embryonic brain growth, high levels of Lgi1 expression were found in the surface ectoderm, the neuroepithelium, mesenchymal connective tissue, hippocampus, and sensory organs, such as eye, tongue, and the olfactory bulb. Lgi1 was also found in the cranial nerve nuclei and ganglia, such as vestibular, trigeminal, and dorsal ganglia. Expression of Lgi1 followed an orchestrated pattern during mouse development becoming more subdued in areas of the neocortex of the mid- and hind-brain in early postnatal animals, although high expression levels were retained in the choroid plexus and hippocampus. In late postnatal stages, Lgi1 expression continued to be detected in many areas in the brain including, hippocampus, paraventricular thalamic nuclei, inferior colliculus, and the cerebral aqueduct. We also showed that Lgi1-expressing cells co-express nestin, DCX, and beta-III tubulin suggesting that Lgi1-expressing cells are migratory neuroblasts.These observations imply that Lgi1 may have a role in establishing normal brain architecture and neuronal functions during brain development suggesting that it may be involved in neurogenesis and neuronal plasticity, which become more specifically defined in the adult animal. Testo completo dell'articolo | | 21569517
|
BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization.
Hitt, BD; Jaramillo, TC; Chetkovich, DM; Vassar, R
Molecular neurodegeneration
5
31
2009
Mostra il sommario
BACE1 is a key enzyme in the generation of the Abeta peptide that plays a central role in the pathogenesis of Alzheimer's disease. While BACE1 is an attractive therapeutic target, its normal physiological function remains largely unknown. Examination of BACE1-/- mice can provide insight into this function and also help anticipate consequences of BACE1 inhibition. Here we report a seizure-susceptibility phenotype that we have identified and characterized in BACE1-/- mice.We find that electroencephalographic recordings reveal epileptiform abnormalities in some BACE1-/- mice, occasionally including generalized tonic-clonic and absence seizures. In addition, we find that kainic acid injection induces seizures of greater severity in BACE1-/- mice relative to BACE1+/+ littermates, and causes excitotoxic cell death in a subset of BACE1-/- mice. This hyperexcitability phenotype is variable and appears to be manifest in approximately 30% of BACE1-/- mice. Finally, examination of the expression and localization of the voltage-gated sodium channel alpha-subunit Nav1.2 reveals no correlation with BACE1 genotype or any measure of seizure susceptibility.Our data indicate that BACE1 deficiency predisposes mice to spontaneous and pharmacologically-induced seizure activity. This finding has implications for the development of safe therapeutic strategies for reducing Abeta levels in Alzheimer's disease. Further, we demonstrate that altered sodium channel expression and axonal localization are insufficient to account for the observed effect, warranting investigation of alternative mechanisms. | | 20731874
|