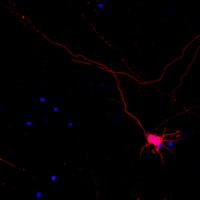
Tau pathogenesis is promoted by Aβ1-42 but not Aβ1-40.
Hu, X; Li, X; Zhao, M; Gottesdiener, A; Luo, W; Paul, S
Molecular neurodegeneration
9
52
2014
Mostra il sommario
The relationship between the pathogenic amyloid β-peptide species Aβ1-42 and tau pathology has been well studied and suggests that Aβ1-42 can accelerate tau pathology in vitro and in vivo. The manners if any in which Aβ1-40 interacts with tau remains poorly understood. In order to answer this question, we used cell-based system, transgenic fly and transgenic mice as models to study the interaction between Aβ1-42 and Aβ1-40.In our established cellular model, live cell imaging (using confocal microscopy) combined with biochemical data showed that exposure to Aβ1-42 induced cleavage, phosphorylation and aggregation of wild-type/full length tau while exposure to Aβ1-40 didn't. Functional studies with Aβ1-40 were carried out in tau-GFP transgenic flies and showed that Aβ1-42, as previously reported, disrupted cytoskeletal structure while Aβ1-40 had no effect at same dose. To further explore how Aβ1-40 affects tau pathology in vivo, P301S mice (tau transgenic mice) were injected intracerebrally with either Aβ1-42 or Aβ1-40. We found that treatment with Aβ1-42 induced tau phosphorylation, cleavage and aggregation of tau in P301S mice. By contrast, Aβ1-40 injection didn't alter total tau, phospho-tau (recognized by PHF-1) or cleavage of tau, but interestingly, phosphorylation at Ser262 was shown to be significantly decreased after direct inject of Aβ1-40 into the entorhinal cortex of P301S mice.These results demonstrate that Aβ1-40 plays different role in tau pathogenesis compared to Aβ1-42. Aβ1-40 may have a protective role in tau pathogenesis by reducing phosphorylation at Ser262, which has been shown to be neurotoxic. | | | 25417177
 |
Increased tau phosphorylation and tau truncation, and decreased synaptophysin levels in mutant BRI2/tau transgenic mice.
Garringer, HJ; Murrell, J; Sammeta, N; Gnezda, A; Ghetti, B; Vidal, R
PloS one
8
e56426
2013
Mostra il sommario
Familial Danish dementia (FDD) is an autosomal dominant neurodegenerative disease caused by a 10-nucleotide duplication-insertion in the BRI(2) gene. FDD is clinically characterized by loss of vision, hearing impairment, cerebellar ataxia and dementia. The main neuropathologic findings in FDD are the deposition of Danish amyloid (ADan) and the presence of neurofibrillary tangles (NFTs). Here we investigated tau accumulation and truncation in double transgenic (Tg-FDD-Tau) mice generated by crossing transgenic mice expressing human Danish mutant BRI(2) (Tg-FDD) with mice expressing human 4-repeat mutant Tau-P301S (Tg-Tau). Compared to Tg-Tau mice, we observed a significant enhancement of tau deposition in Tg-FDD-Tau mice. In addition, a significant increase in tau cleaved at aspartic acid (Asp) 421 was observed in Tg-FDD-Tau mice. Tg-FDD-Tau mice also showed a significant decrease in synaptophysin levels, occurring before widespread deposition of fibrillar ADan and tau can be observed. Thus, the presence of soluble ADan/mutant BRI(2) can lead to significant changes in tau metabolism and synaptic dysfunction. Our data provide new in vivo insights into the pathogenesis of FDD and the pathogenic pathway(s) by which amyloidogenic peptides, regardless of their primary amino acid sequence, can cause neurodegeneration. | Immunohistochemistry | | 23418567
|
Apoptosis in transgenic mice expressing the P301L mutated form of human tau.
Rita M Ramalho, Ricardo J S Viana, Rui E Castro, Clifford J Steer, Walter C Low, Cecília M P Rodrigues
Molecular medicine (Cambridge, Mass.)
14
309-17
2008
Mostra il sommario
The rTg4510 mouse is a tauopathy model, characterized by massive neurodegeneration in Alzheimer's disease (AD)-relevant cortical and limbic structures, deficits in spatial reference memory, and progression of neurofibrillary tangles (NFT). In this study, we examined the role of apoptosis in neuronal loss and associated tau pathology. The results showed that DNA fragmentation and caspase-3 activation are common in the hippocampus and frontal cortex of young rTg4510 mice. These changes were associated with cleavage of tau into smaller intermediate fragments, which persist with age. Interestingly, active caspase-3 was often co-localized with cleaved tau. In vitro, fibrillar Abeta(1-42) resulted in nuclear fragmentation, caspase activation, and caspase-3-induced cleavage of tau. Notably, incubation with the antiapoptotic molecule tauroursodeoxycholic acid abrogated apoptosis-mediated cleavage of tau in rat cortical neurons. In conclusion, caspase-3-cleaved intermediate tau species occurred early in rTg54510 brains and preceded cell loss in Abeta-exposed cultured neurons. These results suggest a potential role of apoptosis in neurodegeneration. Testo completo dell'articolo | | | 18368144
|
Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease.
Guillozet-Bongaarts, Angela L, et al.
Neurobiol. Aging, 26: 1015-22 (2005)
2004
Mostra il sommario
The microtubule-associated protein, tau, is a highly soluble molecule that is nonetheless capable of self-association into filamentous deposits characteristic of a number of neurodegenerative diseases. This state change is thought to be driven by phosphorylation and/or C-terminal truncation events resulting in intracellular inclusions, such as the neurofibrillary tangles (NFTs) in Alzheimer's disease (AD). Previously, we reported the existence of a novel truncation event, cleavage at aspartic acid(421), presumably by a caspase, and also described a monoclonal antibody (Tau-C3) specific for tau cleaved at this site. Here, we report the timing of this cleavage event relative to other antibody-targeted alterations in the tau molecule during the course of NFT evolution in AD. Immunohistochemical studies indicate that cleavage at aspartic acid(421) occurs after formation of the Alz50 epitope but prior to formation of the Tau-66 epitope and truncation at glutamic acid(391) (formation of the MN423 epitope). Thus, creation of the Tau-C3 epitope appears to occur relatively early in the disease state, contemporaneous with the initial Alz50 folding event that heralds the appearance of filamentous tau in NFTs, neuropil threads, and the dystrophic neurites surrounding amyloid plaques. | | Human | 15748781
|
The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration.
Park, So-Young and Ferreira, Adriana
J. Neurosci., 25: 5365-75 (2005)
2004
Mostra il sommario
Recently, we have shown that the microtubule-associated protein tau is essential for beta-amyloid (Abeta)-induced neurotoxicity in hippocampal neurons. However, the mechanisms by which tau mediates Abeta-induced neurite degeneration remain poorly understood. In the present study, we analyzed whether tau cleavage played a role in these events. Our results showed that pre-aggregated Abeta induced the generation of a 17 kDa tau fragment in cultured hippocampal neurons. The generation of this fragment was preceded by the activation of calpain-1. Conversely, inhibitors of this protease, but not of caspases, completely prevented tau proteolysis leading to the generation of the 17 kDa fragment and significantly reduced Abeta-induced neuronal death. Furthermore, the expression of this fragment in cultured hippocampal neurons induced the formation of numerous varicosity-bearing tortuous processes, as well as the complete degeneration of some of those neurite processes. These results suggest that Abeta-induced neurotoxicity may be mediated, at least in part, through the calpain-mediated generation of a toxic 17 kDa tau fragment. Collectively, these results provide insight into a novel mechanism by which tau could mediate Abeta-induced neurotoxicity. | | | 15930385
|
Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease.
Horowitz, Peleg M, et al.
J. Neurosci., 24: 7895-902 (2004)
2004
Mostra il sommario
Alzheimer's disease (AD) is a progressive amnestic dementia that involves post-translational hyperphosphorylation, enzymatic cleavage, and conformational alterations of the microtubule-associated protein tau. The truncation state of tau influences many of its pathologic characteristics, including its ability to assume AD-related conformations and to assemble into filaments. Cleavage also appears to be an important marker in AD progression. Although C-terminal truncation of tau at D421 has recently been attributed to the apoptotic enzyme caspase-3, N-terminal processing of the protein remains mostly uncharacterized. Here, we report immunohistochemical staining in a cohort of 35 cases ranging from noncognitively impaired to early AD with a panel of three N-terminal anti-tau antibodies: Tau-12, 5A6, and 9G3-pY18. Of these three, the phosphorylation-independent epitope of 5A6 was the earliest to emerge in the pathological lesions of tau, followed by the appearance of the Tau-12 epitope. The unmasking of the Tau-12 epitope in more mature 5A6-positive tangles was not correlated with tau phosphorylation at tyrosine 18 (9G3-pY18). Still, later in the course of tangle evolution, the extreme N terminus of tau was lost, correlating temporally with the appearance of a C-terminal caspase-truncated epitope lacking residues 422-441. In addition, caspase-6 cleaved the N terminus of tau in vitro, preventing immunoreactivity with both Tau-12 and 5A6. Mass spectrometry confirmed that the in vitro caspase-6 truncation site is D13, a semicanonical and hitherto undescribed caspase cleavage site in tau. Collectively, these results suggest a role for caspase-6 and N-terminal truncation of tau during neurofibrillary tangle evolution and the progression of Alzheimer's disease. | | | 15356202
|
Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease.
Gamblin, T Chris, et al.
Proc. Natl. Acad. Sci. U.S.A., 100: 10032-7 (2003)
2003
Mostra il sommario
The principal pathological features of Alzheimer's disease (AD) are extracellular amyloid plaques and intracellular neurofibrillary tangles, the latter composed of the microtubule-binding protein tau assembled into paired helical and straight filaments. Recent studies suggest that these pathological entities may be functionally linked, although the mechanisms by which amyloid deposition promotes pathological tau filament assembly are poorly understood. Here, we report that tau is proteolyzed by multiple caspases at a highly conserved aspartate residue (Asp421) in its C terminus in vitro and in neurons treated with amyloid-beta (Abeta) (1-42) peptide. Tau is rapidly cleaved at Asp421 in Abeta-treated neurons (within 2 h), and its proteolysis appears to precede the nuclear events of apoptosis. We also demonstrate that caspase cleavage of tau generates a truncated protein that lacks its C-terminal 20 amino acids and assembles more rapidly and more extensively into tau filaments in vitro than wild-type tau. Using a monoclonal antibody that specifically recognizes tau truncated at Asp421, we show that tau is proteolytically cleaved at this site in the fibrillar pathologies of AD brain. Taken together, our results suggest a novel mechanism linking amyloid deposition and neurofibrillary tangles in AD: Abeta peptides promote pathological tau filament assembly in neurons by triggering caspase cleavage of tau and generating a proteolytic product with enhanced polymerization kinetics. | | | 12888622
|