Neuropathologic analysis of Tyr69His TTR variant meningovascular amyloidosis with dementia.
Ziskin, JL; Greicius, MD; Zhu, W; Okumu, AN; Adams, CM; Plowey, ED
Acta neuropathologica communications
3
43
2015
Mostra il sommario
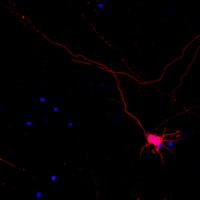
Transthyretin/TTR gene mutations usually cause systemic amyloidotic diseases. Few TTR variants preferentially affect the central nervous system, manifesting as oculoleptomeningeal amyloidosis. Patients with TTR meningovascular amyloidosis often show dementia, however the neuropathologic features of dementia in these cases have not been elucidated. We report the neuropathologic findings from a brain autopsy of a 72-year-old man with the rare Tyr69His (Y69H) TTR gene variant, dementia and ataxia. Severe amyloid deposits were observed in the leptomeninges and in a subpial and subependymal distribution. Mass spectrometry analysis demonstrated that the amyloid deposits were comprised of over 80 % of the variant TTR. TTR was undetectable by mass spectrometry in the neocortex subjacent to the subpial amyloid deposits. Subpial TTR amyloid deposits were associated with brisk superficial reactive gliosis and siderosis in the neocortex and cerebellar cortex. Subependymal TTR amyloid deposits were associated with subjacent myelin pallor in the hippocampal outflow tract structures including the alveus, fimbria and fornix. Phospho-tau immunostains demonstrated transentorhinal-stage neurofibrillary degeneration (Braak stage II) which, in the absence of neocortical amyloid-beta and neuritic plaques, was indicative of primary age-related tauopathy (PART). However, distinctive phospho-tau aggregates were observed subjacent to the subpial TTR amyloid deposits in all regions of the neocortex, including the primary motor and striate cortices, suggesting a potential link between TTR amyloid and neocortical tauopathy. Our report reveals novel insights into the potential neuropathologic substrates of dementia in variant TTR amyloidosis that need to be investigated in larger autopsy series. | 26156087
 |
Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus.
Liu, C; Götz, J
PloS one
8
e84849
2013
Mostra il sommario
In the adult murine brain, the microtubule-associated protein tau exists as three major isoforms, which have four microtubule-binding repeats (4R), with either no (0N), one (1N) or two (2N) amino-terminal inserts. The human brain expresses three additional isoforms with three microtubule-binding repeats (3R) each. However, little is known about the role of the amino-terminal inserts and how the 0N, 1N and 2N tau species differ. In order to investigate this, we generated a series of isoform-specific antibodies and performed a profiling by Western blotting and immunohistochemical analyses using wild-type mice in three age groups: two months, two weeks and postnatal day 0 (P0). This revealed that the brain is the only organ to express tau at significant levels, with 0N4R being the predominant isoform in the two month-old adult. Subcellular fractionation of the brain showed that the 1N isoform is over-represented in the soluble nuclear fraction. This is in agreement with the immunohistochemical analysis as the 1N isoform strongly localizes to the neuronal nucleus, although it is also found in cell bodies and dendrites, but not axons. The 0N isoform is mainly found in cell bodies and axons, whereas nuclei and dendrites are only slightly stained with the 0N antibody. The 2N isoform is highly expressed in axons and in cell bodies, with a detectable expression in dendrites and a very slight expression in nuclei. The 2N isoform that was undetectable at P0, in adult brain was mainly found localized to cell bodies and dendrites. Together these findings reveal significant differences between the three murine tau isoforms that are likely to reflect different neuronal functions. | 24386422
|
Parenchymal and vascular lesions in ageing equine brains: histological and immunohistochemical studies.
M T Capucchio,M Márquez,P Pregel,L Foradada,M Bravo,G Mattutino,C Torre,D Schiffer,D Catalano,F Valenza,F Guarda,M Pumarola
Journal of comparative pathology
142
2009
Mostra il sommario
Many age-related changes are described in the nervous system of different species, but detailed studies of brain lesions in ageing horses are lacking. The aim of the present study was to systematically characterize lesions in the brains of 60 horses aged from 7 to 23 years. No gross changes were present in any brain. Microscopically, spongiform changes, lipofuscin storage, corpora amylacea, gliosis and satellitosis were common, together with axonal and neuronal swellings. The most important findings were the presence of pseudocalcium-calcium (pCa-Ca) deposits and arterial wall degeneration. Scanning electron microscopical examination of two cases with vascular mineralization revealed marked deposition of an amorphous substance in the vessel walls that was probably formed by a polyanionic protein matrix and a mineral component. Immunohistochemically, numerous axonal spheroids were positively labelled for ubiquitin. No PrPsc was detected in sections with neuronal vacuolation. Neuronal swelling, corpora amylacea, hippocampal Tau-positive neurons and methenamine-positive diffuse (preamyloid) plaques were also detected. Congo red staining failed to detect amyloid deposition. The characterization of age-related lesions in the brains of these horses will allow these changes to be discriminated from pathological processes in future studies. Some lesions described here, including some vascular changes, the presence of diffuse plaques and tau accumulation in hippocampal neurons, have not been described previously in the horse. | 19744668
|
Differential involvement and heterogeneous phosphorylation of tau isoforms in progressive supranuclear palsy.
Gibb, G M, et al.
Brain Res. Mol. Brain Res., 121: 95-101 (2004)
2004
Mostra il sommario
We found previously that aggregated insoluble tau protein in progressive supranuclear palsy (PSP) brains exhibits a heterogeneous pattern that is not segregated by the type of clinical presentation. Here we have investigated tau isoform composition from 20 PSP cases and found marked variation between different brains. Cases were classified into three groups, each comprising essentially of (1) 1N4R; (2) 1N4R and 1N3R; or (3) 1N4R, 1N3R and 0N4R tau isoforms. There was also an absence of a simple relationship between isoform composition and the pattern of insoluble tau before dephosphorylation. We conclude that there is distinct molecular heterogeneity in the involvement of tau isoforms in the tau pathology in PSP. | 14969740
|
Dementia with Lewy bodies from the perspective of tauopathy.
Iseki, Eizo, et al.
Acta Neuropathol., 105: 265-70 (2003)
2003
Mostra il sommario
We immunohistochemically investigated the prevalence and pattern of phosphorylated tau accumulation in neurons and glia in 46 cases of dementia with Lewy bodies (DLB). Tau-positive neurons composed of neurofibrillary tangles (NFT) and pretangle neurons were found in the hippocampal area in all 46 cases, although the ratio of pretangle neurons in tau-positive neurons was higher in the cases showing low NFT stages. Tau-positive astrocytes were found in the periventricular area in 18 of 46 cases, and partly represented argyrophilic thorn-shaped astrocytes. In contrast, tau-positive oligodendroglia were found in the subcortical white matter in 9 of 46 cases, and represented argyrophilic coiled bodies. Tau-positive argyrophilic grains were found in the hippocampal area in the same cases as those with coiled bodies. The 9 cases with tau-positive coiled bodies and grains were included in the 18 cases with tau-positive astrocytes, and showed larger proportions in the low NFT stages than the 46 cases with tau-positive neurons. Tau-positive neurons were positive both to anti-three-repeat (3R) and -4R tau-specific antibodies, while tau-positive astrocytes, coiled bodies and grains were predominantly positive to anti-4R tau-specific antibody. These tau-positive structures were negative to anti-alpha-synuclein antibody. These findings suggest that the tau accumulation in DLB represents both tau-positive neurons with all six tau isoforms and tau-positive astrocytes, coiled bodies and grains with the 4R tau isoform, and that the different cytoskeletal abnormalities form a link between some neurodegenerative dementing disorders including DLB. | 12557014
|
The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy.
Hogg, Marion, et al.
Acta Neuropathol., 106: 323-36 (2003)
2003
Mostra il sommario
We report a case of rapidly progressive frontotemporal dementia presenting at age 33 years. At autopsy there was severe atrophy of the frontal and temporal lobes. Tau-positive Pick bodies, which ultrastructurally were composed of straight filaments, were present, accompanied by severe neuronal loss and gliosis. RD3, a tau antibody specific for the three-repeat (3R) isoforms, labeled the Pick bodies. ET3, a four-repeat (4R) isoform-specific tau antibody, did not label Pick bodies, but highlighted rare astrocytes, and threads in white matter bundles in the corpus striatum. Analysis of the tau gene revealed an L266V mutation in exon 9. Analysis of brain tissue from this case revealed elevated levels of exon 10+ tau RNA and soluble 4R tau. However, both 3R and 4R isoforms were present in sarkosyl-insoluble tau fractions with a predominance of the shortest 3R isoform. The L266V mutation is associated with decreased rate and extent of tau-induced microtubule assembly, and a 3R isoform-specific increase in tau self assembly as measured by an in vitro assay. Combined, these data indicate that L266V is a pathogenic tau mutation that is associated with Pick-like pathology. In addition, the results of the RD3 and ET3 immunostains clearly explain for the first time the presence of both 3R and 4R tau isoforms in preparations of insoluble tau from some Pick's disease cases. | 12883828
|
Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies.
de Silva, R, et al.
Neuropathol. Appl. Neurobiol., 29: 288-302 (2003)
2003
Mostra il sommario
Pathological inclusions containing fibrillar aggregates of hyperphosphorylated tau protein are a characteristic feature in the tauopathies, which include Alzheimer's disease, frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Tau isoform composition and cellular and regional distribution as well as morphology of these inclusions vary in each disorder. Recently, several pathological missense and exon 10 splice-donor site mutations of the tau gene were identified in FTDP-17. Exon 10 codes for the second of four microtubule-binding repeat domains. The splice-site mutations result in increased inclusion of exon 10 which causes a relative increase in tau isoforms containing four microtubule-binding repeat domains over those containing three repeat domains. This could be a central aetiological mechanism in FTDP-17 and, perhaps, other related tauopathies. We have investigated changes in the ratio and distribution of three-repeat and four-repeat tau in the different tauopathies as a basis of the phenotypic range of these disorders and the selective vulnerability of different subsets of neurones. In this study, we have developed two monoclonal antibodies, RD3 and RD4 that effectively distinguish these closely related tau isoforms. These new isoform-specific antibodies are useful tools for analysing tau isoform expression and distribution as well as pathological changes in the human brain. | 12787326
|
Argyrophilic grain disease is a sporadic 4-repeat tauopathy
Togo, Takashi, et al
J Neuropathol Exp Neurol, 61:547-56 (2002)
2002
| 12071638
|
The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons.
Utton, Michelle A, et al.
J. Neurosci., 22: 6394-400 (2002)
2002
Mostra il sommario
We demonstrate that the microtubule-associated protein tau, in the form of enhanced green fluorescent protein (EGFP) tau, is transported along axons of neurons in culture in the slow component of axonal transport with a speed comparable with that previously measured in vivo. It was demonstrated that the EGFP tag has no effect on transport characteristics, and the methodology enables slow transport rates of individual tau isoforms and tau mutants to be measured. We also expressed EGFP-tagged tau isoforms containing either three or four C-terminal repeats and zero or two N-terminal inserts in cultured neurons. No significant differences were found in the average rate of slow transport of the wild-type tau isoforms, suggesting that the exon 10 C-terminal repeat or the N-terminal inserts do not contain regions that play a significant regulatory role in axonal transport. Similarly, we found that missense mutations in tau have no noticeable effect on the rate of transport; hence their ability to cause neurodegeneration is by another mechanism other than that affecting the overall slow axonal transport of tau. | 12151518
|
The complex relationship between soluble and insoluble tau in tauopathies revealed by efficient dephosphorylation and specific antibodies
Hanger, D P, et al
FEBS Lett, 531:538-42 (2002)
2002
| 12435607
|