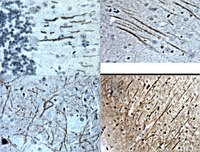
VEGF induces sensory and motor peripheral plasticity, alters bladder function, and promotes visceral sensitivity.
Malykhina, AP; Lei, Q; Erickson, CS; Epstein, ML; Saban, MR; Davis, CA; Saban, R
BMC physiology
12
15
2011
Mostra il sommario
This work tests the hypothesis that bladder instillation with vascular endothelial growth factor (VEGF) modulates sensory and motor nerve plasticity, and, consequently, bladder function and visceral sensitivity.In addition to C57BL/6J, ChAT-cre mice were used for visualization of bladder cholinergic nerves. The direct effect of VEGF on the density of sensory nerves expressing the transient receptor potential vanilloid subfamily 1 (TRPV1) and cholinergic nerves (ChAT) was studied one week after one or two intravesical instillations of the growth factor.To study the effects of VEGF on bladder function, mice were intravesically instilled with VEGF and urodynamic evaluation was assessed. VEGF-induced alteration in bladder dorsal root ganglion (DRG) neurons was performed on retrogradly labeled urinary bladder afferents by patch-clamp recording of voltage gated Na+ currents. Determination of VEGF-induced changes in sensitivity to abdominal mechanostimulation was performed by application of von Frey filaments.In addition to an overwhelming increase in TRPV1 immunoreactivity, VEGF instillation resulted in an increase in ChAT-directed expression of a fluorescent protein in several layers of the urinary bladder. Intravesical VEGF caused a profound change in the function of the urinary bladder: acute VEGF (1 week post VEGF treatment) reduced micturition pressure and longer treatment (2 weeks post-VEGF instillation) caused a substantial reduction in inter-micturition interval. In addition, intravesical VEGF resulted in an up-regulation of voltage gated Na(+) channels (VGSC) in bladder DRG neurons and enhanced abdominal sensitivity to mechanical stimulation.For the first time, evidence is presented indicating that VEGF instillation into the mouse bladder promotes a significant increase in peripheral nerve density together with alterations in bladder function and visceral sensitivity. The VEGF pathway is being proposed as a key modulator of neural plasticity in the pelvis and enhanced VEGF content may be associated with visceral hyperalgesia, abdominal discomfort, and/or pelvic pain. | 23249422
 |
VEGF signaling mediates bladder neuroplasticity and inflammation in response to BCG.
Saban, MR; Davis, CA; Avelino, A; Cruz, F; Maier, J; Bjorling, DE; Sferra, TJ; Hurst, RE; Saban, R
BMC physiology
11
16
2010
Mostra il sommario
This work tests the hypothesis that increased levels of vascular endothelial growth factor (VEGF) observed during bladder inflammation modulates nerve plasticity.Chronic inflammation was induced by intravesical instillations of Bacillus Calmette-Guérin (BCG) into the urinary bladder and the density of nerves expressing the transient receptor potential vanilloid subfamily 1 (TRPV1) or pan-neuronal marker PGP9.5 was used to quantify alterations in peripheral nerve plasticity. Some mice were treated with B20, a VEGF neutralizing antibody to reduce the participation of VEGF. Additional mice were treated systemically with antibodies engineered to specifically block the binding of VEGF to NRP1 (anti-NRP1B) and NRP2 (NRP2B), or the binding of semaphorins to NRP1 (anti-NRP1 A) to diminish activity of axon guidance molecules such as neuropilins (NRPs) and semaphorins (SEMAs). To confirm that VEGF is capable of inducing inflammation and neuronal plasticity, another group of mice was instilled with recombinant VEGF165 or VEGF121 into the urinary bladder.The major finding of this work was that chronic BCG instillation resulted in inflammation and an overwhelming increase in both PGP9.5 and TRPV1 immunoreactivity, primarily in the sub-urothelium of the urinary bladder. Treatment of mice with anti-VEGF neutralizing antibody (B20) abolished the effect of BCG on inflammation and nerve density.NRP1A and NRP1B antibodies, known to reduce BCG-induced inflammation, failed to block BCG-induced increase in nerve fibers. However, the NRP2B antibody dramatically potentiated the effects of BCG in increasing PGP9.5-, TRPV1-, substance P (SP)-, and calcitonin gene-related peptide (CGRP)-immunoreactivity (IR). Finally, instillation of VEGF121 or VEGF165 into the mouse bladder recapitulated the effects of BCG and resulted in a significant inflammation and increase in nerve density.For the first time, evidence is being presented supporting that chronic BCG instillation into the mouse bladder promotes a significant increase in peripheral nerve density that was mimicked by VEGF instillation. Effects of BCG were abolished by pre-treatment with neutralizing VEGF antibody. The present results implicate the VEGF pathway as a key modulator of inflammation and nerve plasticity, introduces a new animal model for investigation of VEGF-induced nerve plasticity, and suggests putative mechanisms underlying this phenomenon. | 22059553
|
Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells.
Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L
RNA
16
324-37. Epub 2009 Dec 21.
2009
Mostra il sommario
The genetic networks controlling stem cell identity are the focus of intense interest, due to their obvious therapeutic potential as well as exceptional relevance to models of early development. Genome-wide mapping of transcriptional networks in mouse embryonic stem cells (mESCs) reveals that many endogenous noncoding RNA molecules, including long noncoding RNAs (lncRNAs), may play a role in controlling the pluripotent state. We performed a genome-wide screen that combined full-length mESC transcriptome genomic mapping data with chromatin immunoprecipitation genomic location maps of the key mESC transcription factors Oct4 and Nanog. We henceforth identified four mESC-expressed, conserved lncRNA-encoding genes residing proximally to active genomic binding sites of Oct4 and Nanog. Accordingly, these four genes have potential roles in pluripotency. We show that two of these lncRNAs, AK028326 (Oct4-activated) and AK141205 (Nanog-repressed), are direct targets of Oct4 and Nanog. Most importantly, we demonstrate that these lncRNAs are not merely controlled by mESC transcription factors, but that they themselves regulate developmental state: knockdown and overexpression of these transcripts lead to robust changes in Oct4 and Nanog mRNA levels, in addition to alterations in cellular lineage-specific gene expression and in the pluripotency of mESCs. We further characterize AK028326 as a co-activator of Oct4 in a regulatory feedback loop. These results for the first time implicate lncRNAs in the modulation of mESC pluripotency and expand the established mESC regulatory network model to include functional lncRNAs directly controlled by key mESC transcription factors. Testo completo dell'articolo | 20026622
|
Serotonin receptor 1A-modulated phosphorylation of glycine receptor α3 controls breathing in mice.
Manzke, T; Niebert, M; Koch, UR; Caley, A; Vogelgesang, S; Hülsmann, S; Ponimaskin, E; Müller, U; Smart, TG; Harvey, RJ; Richter, DW
The Journal of clinical investigation
120
4118-28
2009
Mostra il sommario
Rhythmic breathing movements originate from a dispersed neuronal network in the medulla and pons. Here, we demonstrate that rhythmic activity of this respiratory network is affected by the phosphorylation status of the inhibitory glycine receptor α3 subtype (GlyRα3), which controls glutamatergic and glycinergic neuronal discharges, subject to serotonergic modulation. Serotonin receptor type 1A-specific (5-HTR1A-specific) modulation directly induced dephosphorylation of GlyRα3 receptors, which augmented inhibitory glycine-activated chloride currents in HEK293 cells coexpressing 5-HTR1A and GlyRα3. The 5-HTR1A-GlyRα3 signaling pathway was distinct from opioid receptor signaling and efficiently counteracted opioid-induced depression of breathing and consequential apnea in mice. Paradoxically, this rescue of breathing originated from enhanced glycinergic synaptic inhibition of glutamatergic and glycinergic neurons and caused disinhibition of their target neurons. Together, these effects changed respiratory phase alternations and ensured rhythmic breathing in vivo. GlyRα3-deficient mice had an irregular respiratory rhythm under baseline conditions, and systemic 5-HTR1A activation failed to remedy opioid-induced respiratory depression in these mice. Delineation of this 5-HTR1A-GlyRα3 signaling pathway offers a mechanistic basis for pharmacological treatment of opioid-induced apnea and other breathing disturbances caused by disorders of inhibitory synaptic transmission, such as hyperekplexia, hypoxia/ischemia, and brainstem infarction. | 20978350
|
Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of mu-opioid receptors in CNS neurons.
Yu, YJ; Arttamangkul, S; Evans, CJ; Williams, JT; von Zastrow, M
The Journal of neuroscience : the official journal of the Society for Neuroscience
29
222-33
2009
Mostra il sommario
mu-Opioid receptors (MORs) are G-protein-coupled receptors (GPCRs) that mediate the physiological effects of endogenous opioid neuropeptides and opiate drugs such as morphine. MORs are coexpressed with neurokinin 1 receptors (NK1Rs) in several regions of the CNS that control opioid dependence and reward. NK1R activation affects opioid reward specifically, however, and the cellular basis for this specificity is unknown. We found that ligand-induced activation of NK1Rs produces a cell-autonomous and nonreciprocal inhibition of MOR endocytosis induced by diverse opioids. Studies using epitope-tagged receptors expressed in cultured striatal neurons and a neuroblastoma cell model indicated that this heterologous regulation is mediated by NK1R-dependent sequestration of arrestins on endosome membranes. First, endocytic inhibition mediated by wild-type NK1Rs was overcome in cells overexpressing beta-arrestin2, a major arrestin isoform expressed in striatum. Second, NK1R activation promoted sequestration of beta-arrestin2 on endosomes, whereas MOR activation did not. Third, heterologous inhibition of MOR endocytosis was prevented by mutational disruption of beta-arrestin2 sequestration by NK1Rs. NK1R-mediated regulation of MOR trafficking was associated with reduced opioid-induced desensitization of adenylyl cyclase signaling in striatal neurons. Furthermore, heterologous regulation of MOR trafficking was observed in both amygdala and locus ceruleus neurons that naturally coexpress these receptors. These results identify a cell-autonomous mechanism that may underlie the highly specific effects of NK1R on opioid signaling and suggest, more generally, that receptor-specific trafficking of arrestins may represent a fundamental mechanism for coordinating distinct GPCR-mediated signals at the level of individual CNS neurons. | 19129399
|