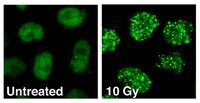
TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution.
Broderick, R; Nieminuszczy, J; Blackford, AN; Winczura, A; Niedzwiedz, W
Nature communications
6
6572
2015
Mostra il sommario
During mitosis, sister chromatids must be faithfully segregated to ensure that daughter cells receive one copy of each chromosome. However, following replication they often remain entangled. Topoisomerase IIα (TOP2A) has been proposed to resolve such entanglements, but the mechanisms governing TOP2A recruitment to these structures remain poorly understood. Here, we identify TOPBP1 as a novel interactor of TOP2A, and reveal that it is required for TOP2A recruitment to ultra-fine anaphase bridges (UFBs) in mitosis. The C-terminal region of TOPBP1 interacts with TOP2A, and TOPBP1 recruitment to UFBs requires its BRCT domain 5. Depletion of TOPBP1 leads to accumulation of UFBs, the majority of which arise from centromeric loci. Accordingly, expression of a TOPBP1 mutant that is defective in TOP2A binding phenocopies TOP2A depletion. These findings provide new mechanistic insights into how TOP2A promotes resolution of UFBs during mitosis, and highlights a pivotal role for TOPBP1 in this process. | 25762097
 |
BRCA1 pathway function in basal-like breast cancer cells.
Hill, SJ; Clark, AP; Silver, DP; Livingston, DM
Molecular and cellular biology
34
3828-42
2014
Mostra il sommario
Sporadic basal-like cancers (BLCs) are a common subtype of breast cancer that share multiple biological properties with BRCA1-mutated breast tumors. Despite being BRCA1(+/+), sporadic BLCs are widely viewed as phenocopies of BRCA1-mutated breast cancers, because they are hypothesized to manifest a BRCA1 functional defect or breakdown of a pathway(s) in which BRCA1 plays a major role. The role of BRCA1 in the repair of double-strand DNA breaks by homologous recombination (HR) is its best understood function and the function most often implicated in BRCA1 breast cancer suppression. Therefore, it is suspected that sporadic BLCs exhibit a defect in HR. To test this hypothesis, multiple DNA damage repair assays focused on several types of repair were performed on a group of cell lines classified as sporadic BLCs and on controls. The sporadic BLC cell lines failed to exhibit an overt HR defect. Rather, they exhibited defects in the repair of stalled replication forks, another BRCA1 function. These results provide insight into why clinical trials of poly(ADP-ribose) polymerase (PARP) inhibitors, which require an HR defect for efficacy, have been unsuccessful in sporadic BLCs, unlike cisplatin, which elicits DNA damage that requires stalled fork repair and has shown efficacy in sporadic BLCs. | 25092866
|
FANC pathway promotes UV-induced stalled replication forks recovery by acting both upstream and downstream Polη and Rev1.
Renaud, E; Rosselli, F
PloS one
8
e53693
2013
Mostra il sommario
To cope with ultraviolet C (UVC)-stalled replication forks and restart DNA synthesis, cells either undergo DNA translesion synthesis (TLS) by specialised DNA polymerases or tolerate the lesions using homologous recombination (HR)-based mechanisms. To gain insight into how cells manage UVC-induced stalled replication forks, we analysed the molecular crosstalk between the TLS DNA polymerases Polη and Rev1, the double-strand break repair (DSB)-associated protein MDC1 and the FANC pathway. We describe three novel functional interactions that occur in response to UVC-induced DNA lesions. First, Polη and Rev1, whose optimal expression and/or relocalisation depend on the FANC core complex, act upstream of FANCD2 and are required for the proper relocalisation of monoubiquitinylated FANCD2 (Ub-FANCD2) to subnuclear foci. Second, during S-phase, Ub-FANCD2 and MDC1 relocalise to UVC-damaged nuclear areas or foci simultaneously but independently of each other. Third, Ub-FANCD2 and MDC1 are independently required for optimal BRCA1 relocalisation. While RPA32 phosphorylation (p-RPA32) and RPA foci formation were reduced in parallel with increasing levels of H2AX phosphorylation and MDC1 foci in UVC-irradiated FANC pathway-depleted cells, MDC1 depletion was associated with increased UVC-induced Ub-FANCD2 and FANCD2 foci as well as p-RPA32 levels and p-RPA32 foci. On the basis of the previous observations, we propose that the FANC pathway participates in the rescue of UVC-stalled replication forks in association with TLS by maintaining the integrity of ssDNA regions and by preserving genome stability and preventing the formation of DSBs, the resolution of which would require the intervention of MDC1. | 23365640
|
Residual dNA and chromosomal damage in ex vivo irradiated blood lymphocytes correlated with late normal tissue response to breast radiotherapy.
Chua ML, Somaiah N, A\'hern R, Davies S, Gothard L, Yarnold J, Rothkamm K
Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology
99
362-6. Epub 2011 Jun 23.
2010
| 21704405
|
Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways.
Rappold, I, et al.
J. Cell Biol., 153: 613-20 (2001)
2001
Mostra il sommario
The tumor suppressor p53 binding protein 1 (53BP1) binds to the DNA-binding domain of p53 and enhances p53-mediated transcriptional activation. 53BP1 contains two breast cancer susceptibility gene 1 COOH terminus (BRCT) motifs, which are present in several proteins involved in DNA repair and/or DNA damage-signaling pathways. Thus, we investigated the potential role of 53BP1 in DNA damage-signaling pathways. Here, we report that 53BP1 becomes hyperphosphorylated and forms discrete nuclear foci in response to DNA damage. These foci colocalize at all time points with phosphorylated H2AX (gamma-H2AX), which has been previously demonstrated to localize at sites of DNA strand breaks. 53BP1 foci formation is not restricted to gamma-radiation but is also detected in response to UV radiation as well as hydroxyurea, camptothecin, etoposide, and methylmethanesulfonate treatment. Several observations suggest that 53BP1 is regulated by ataxia telangiectasia mutated (ATM) after DNA damage. First, ATM-deficient cells show no 53BP1 hyperphosphorylation and reduced 53BP1 foci formation in response to gamma-radiation compared with cells expressing wild-type ATM. Second, wortmannin treatment strongly inhibits gamma-radiation-induced hyperphosphorylation and foci formation of 53BP1. Third, 53BP1 is readily phosphorylated by ATM in vitro. Taken together, these results suggest that 53BP1 is an ATM substrate that is involved early in the DNA damage-signaling pathways in mammalian cells. | 11331310
|