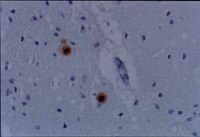
Creutzfeldt-Jakob disease in Mexico.
Leora Velásquez-Pérez, Daniel Rembao-Bojorquez, Jorge Guevara, Rosa María Guadarrama-Torres, Araceli Trejo-Contreras
Neuropathology : official journal of the Japanese Society of Neuropathology
27
419-28
2007
Mostrar resumen
Creutzfeldt-Jakob disease (CJD) is classified within the group of transmissible spongiform encephalopathies (TSE). It is a rapidly progressive illness that affects mental functions. The average age of onset is 50 years. Various tests can help orient the clinical diagnosis, but the confirmatory test is still the post mortem analysis. The aim of this study was to describe the epidemiological, clinical and histopathological characteristics of patients diagnosed as suffering from CJD, at the National Institute of Neurology and Neurosurgery of Mexico (NINN). An observational, descriptive and transversal study was conducted. We collected information concerning these cases from the Departments of Epidemiology and Pathology, as well as the clinical charts of the patients with a diagnosis of CJD. Fifteen cases were registered of which three CJD cases were definite, five probable cases were identified, and seven were possible. The average age of the patients was 49 years. Two definite cases were female and one was male. It is important to improve the systems for surveillance of this type of disease and, furthermore, to permit greater accessibility to laboratories where the procedures necessary for supporting diagnosis can be followed. | 18018474
 |
Molecular and genetic features of a labeled class of spinal substantia gelatinosa neurons in a transgenic mouse.
Adam W Hantman, Edward R Perl
The Journal of comparative neurology
492
90-100
2004
Mostrar resumen
Genetic incorporation in a mouse of a transgene containing the prion promoter and the green fluorescent protein (GFP) coding sequence labels a set of substantia gelatinosa (SG) neurons (SG-GFP) homogenous in morphology, electrophysiology, and gamma-amino-butyric acid expression. In the present analysis the SG-GFP neurons are established to have protein kinase C-betaII immunoreactivity and to lack evidence for the presence of calbindin D-28k, parvalbumin, and protein kinase C-gamma. These neurons were hyperpolarized by mediators of descending control, norepinephrine and serotonin. Sequential polymerase chain reactions established the insertion of the transgene to be in the receptor protein tyrosine phosphatase kappa (RPTP-kappa) and the laminin receptor 1 (ribosomal protein SA) pseudogene 1 locus. RPTP-kappa expression in both GFP-labeled dorsal root ganglia and SG neurons raises the possibility that homophilic interactions of RPTP-kappa contribute to establishment of connections between specific classes of primary afferent and SG neurons. | 16175558
|
Toluidine blue-O staining of prion protein deposits.
A Sánchez, A Guzmán, A Ortiz, D Rembao, B Espinosa, E Zenteno, J Guevara
Histochemistry and cell biology
116
519-24
2001
Mostrar resumen
Prion diseases or transmissible spongiform encephalopathies are a group of fatal neurodegenerative diseases caused by an abnormal form of prion protein (PrP(sc)). In this study, we developed a sensitive histochemical detection of PrP(sc) deposits in a Gertsmann-Sträussler-Scheinker disease (GSS) patient using toluidine blue-O staining, a specific reagent to stain mucins and mucopolysaccharides. Detection of prion deposits correlated with immunohistochemistry using anti-prion antibodies. Control assays were performed using amyloid-beta (Abeta) plaques from Alzheimer's disease (AD) brains. Our results demonstrated that toluidine blue-O staining allowed to recognize 69.1+/-2.6% of the total plaques recognized by the anti-prion antibody. Furthermore, in the 15 studied brain regions from the GSS patient, toluidine blue-O revealed the same recognition pattern as anti-prion labeling. Toluidine blue-O stained specifically the prion deposits but not the Abeta plaques in AD brains. The specificity of the technique was confirmed in a Creutzfeldt-Jakob disease brain. This method opens several possibilities for postmortem diagnoses. Our results also suggest the relevance of specific post-translational modifications of PrP(sc), identified by toluidine blue-O, that might participate in the transformation of PrP(c) to PrP(sc). | 11810193
|
Copper has differential effect on prion protein with polymorphism of position 129.
B S Wong, C Clive, S J Haswell, R A Williamson, D R Burton, P Gambetti, M S Sy, I M Jones, D R Brown
Biochemical and biophysical research communications
269
726-31
1999
Mostrar resumen
The pathology of human prion diseases is affected by polymorphism at amino acid residue 129 of the prion protein gene. Recombinant mouse prion proteins mimicking either form of the polymorphism were prepared to examine their effect on the conformation and the level of superoxide dismutase (SOD) activity of the prion protein. Following the binding of copper atoms to prion protein, antibody mapping and CD analysis detected conformational differences between the two forms of protein. However, neither the level of copper binding nor the level of SOD activity associated with this form of prion protein altered with the identity of codon 129. These results suggest that in the holo-metal binding form of the protein, prion structure but not its SOD activity is affected by polymorphism at codon 129. | 10720484
|
Selective oxidation of methionine residues in prion proteins.
B S Wong, H Wang, D R Brown, I M Jones
Biochemical and biophysical research communications
259
352-5
1998
Mostrar resumen
Prion proteins are central to the pathogenesis of several neurodegenerative diseases through the postulated conversion of the endogenous cellular isoform (PrPc) into a pathogenic isoform (PrPSc). Although the cellular function of normal prion protein remains unresolved a number of studies have shown that prion proteins may be involved in the cellular response to oxidative stress. Here, using purified recombinant sources of mouse and chicken PrP refolded in the presence of copper (II) we show that the methionine residues of the protein are uniquely susceptible to oxidation. We suggest that Met residues may form an essential part of the mechanism of the antioxidant activity exhibited by normal prion protein. | 10362513
|