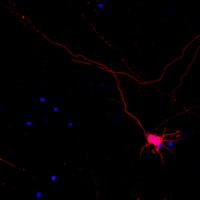
Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer's disease.
Tiwari, SS; d'Orange, M; Troakes, C; Shurovi, BN; Engmann, O; Noble, W; Hortobágyi, T; Giese, KP
Molecular brain
8
6
2015
Mostrar resumen
In Alzheimer's disease synapse loss precedes neuronal loss and correlates best with impaired memory formation. However, the mechanisms underlying synaptic degeneration in Alzheimer's disease are not well known. Further, it is unclear why synapses in AD cerebellum are protected from degeneration. Our recent work on the cyclin-dependent kinase 5 activator p25 suggested that expression of the multifunctional presynaptic molecule cysteine string protein alpha (CSPalpha) may be affected in Alzheimer's disease.Using western blots and immunohistochemistry, we found that CSPalpha expression is reduced in hippocampus and superior temporal gyrus in Alzheimer's disease. Reduced CSPalpha expression occurred before synaptophysin levels drop, suggesting that it contributes to the initial stages of synaptic degeneration. Surprisingly, we also found that CSPalpha expression is upregulated in cerebellum in Alzheimer's disease. This CSPalpha upregulation reached the same level as in young, healthy cerebellum. We tested the idea whether CSPalpha upregulation might be neuroprotective, using htau mice, a model of tauopathy that expresses the entire wild-type human tau gene in the absence of mouse tau. In htau mice CSPalpha expression was found to be elevated at times when neuronal loss did not occur.Our findings provide evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer's disease. In the forebrain CSPalpha expression is reduced early in the disease and this may contribute to the initial stages of synaptic degeneration. In the cerebellum CSPalpha expression is upregulated to young, healthy levels and this may protect cerebellar synapses and neurons to survive. Accordingly, CSPalpha upregulation also occurs in a mouse model of tauopathy only at time when neuronal loss does not take place. | Western Blotting | 25631211
 |
Age-dependent in vivo conversion of mouse cochlear pillar and Deiters' cells to immature hair cells by Atoh1 ectopic expression.
Liu, Z; Dearman, JA; Cox, BC; Walters, BJ; Zhang, L; Ayrault, O; Zindy, F; Gan, L; Roussel, MF; Zuo, J
The Journal of neuroscience : the official journal of the Society for Neuroscience
32
6600-10
2011
Mostrar resumen
Unlike nonmammalian vertebrates, mammals cannot convert inner ear cochlear supporting cells (SCs) into sensory hair cells (HCs) after damage, thus causing permanent deafness. Here, we achieved in vivo conversion of two SC subtypes, pillar cells (PCs) and Deiters' cells (DCs), into HCs by inducing targeted expression of Atoh1 at neonatal and juvenile ages using novel mouse models. The conversion only occurred in ∼10% of PCs and DCs with ectopic Atoh1 expression and started with reactivation of endogenous Atoh1 followed by expression of 11 HC and synaptic markers, a process that took approximately 3 weeks in vivo. These new HCs resided in the outer HC region, formed stereocilia, contained mechanoelectrical transduction channels, and survived for greater than 2 months in vivo; however, they surprisingly lacked prestin and oncomodulin expression and mature HC morphology. In contrast, adult PCs and DCs no longer responded to ectopic Atoh1 expression, even after outer HC damage. Finally, permanent Atoh1 expression in endogenous HCs did not affect prestin expression but caused cell loss of mature HCs. Together, our results demonstrate that in vivo conversion of PCs and DCs into immature HCs by Atoh1 is age dependent and resembles normal HC development. Therefore, combined expression of Atoh1 with additional factors holds therapeutic promise to convert PCs and DCs into functional HCs in vivo for regenerative purposes. | Immunofluorescence | 22573682
|
Deletion of the Ca2+-activated potassium (BK) alpha-subunit but not the BKbeta1-subunit leads to progressive hearing loss.
Rüttiger, L; Sausbier, M; Zimmermann, U; Winter, H; Braig, C; Engel, J; Knirsch, M; Arntz, C; Langer, P; Hirt, B; Müller, M; Köpschall, I; Pfister, M; Münkner, S; Rohbock, K; Pfaff, I; Rüsch, A; Ruth, P; Knipper, M
Proceedings of the National Academy of Sciences of the United States of America
101
12922-7
2004
Mostrar resumen
The large conductance voltage- and Ca2+-activated potassium (BK) channel has been suggested to play an important role in the signal transduction process of cochlear inner hair cells. BK channels have been shown to be composed of the pore-forming alpha-subunit coexpressed with the auxiliary beta1-subunit. Analyzing the hearing function and cochlear phenotype of BK channel alpha-(BKalpha-/-) and beta1-subunit (BKbeta1-/-) knockout mice, we demonstrate normal hearing function and cochlear structure of BKbeta1-/- mice. During the first 4 postnatal weeks also, BKalpha-/- mice most surprisingly did not show any obvious hearing deficits. High-frequency hearing loss developed in BKalpha-/- mice only from approximately 8 weeks postnatally onward and was accompanied by a lack of distortion product otoacoustic emissions, suggesting outer hair cell (OHC) dysfunction. Hearing loss was linked to a loss of the KCNQ4 potassium channel in membranes of OHCs in the basal and midbasal cochlear turn, preceding hair cell degeneration and leading to a similar phenotype as elicited by pharmacologic blockade of KCNQ4 channels. Although the actual link between BK gene deletion, loss of KCNQ4 in OHCs, and OHC degeneration requires further investigation, data already suggest human BK-coding slo1 gene mutation as a susceptibility factor for progressive deafness, similar to KCNQ4 potassium channel mutations. | Immunohistochemistry | 15328414
|