Our broad portfolio consists of multiplex panels that allow you to choose, within the panel, analytes that best meet your needs. On a separate tab you can choose the premixed cytokine format or a single plex kit.
Cell Signaling Kits & MAPmates™
Choose fixed kits that allow you to explore entire pathways or processes. Or design your own kits by choosing single plex MAPmates™, following the provided guidelines.
The following MAPmates™ should not be plexed together:
-MAPmates™ that require a different assay buffer
-Phospho-specific and total MAPmate™ pairs, e.g. total GSK3β and GSK3β (Ser 9)
-PanTyr and site-specific MAPmates™, e.g. Phospho-EGF Receptor and phospho-STAT1 (Tyr701)
-More than 1 phospho-MAPmate™ for a single target (Akt, STAT3)
-GAPDH and β-Tubulin cannot be plexed with kits or MAPmates™ containing panTyr
.
Catalogue Number
Ordering Description
Qty/Pack
List
This item has been added to favorites.
Select A Species, Panel Type, Kit or Sample Type
To begin designing your MILLIPLEX® MAP kit select a species, a panel type or kit of interest.
Custom Premix Selecting "Custom Premix" option means that all of the beads you have chosen will be premixed in manufacturing before the kit is sent to you.
Catalogue Number
Ordering Description
Qty/Pack
List
This item has been added to favorites.
Species
Panel Type
Selected Kit
Qty
Catalogue Number
Ordering Description
Qty/Pack
List Price
96-Well Plate
Qty
Catalogue Number
Ordering Description
Qty/Pack
List Price
Add Additional Reagents (Buffer and Detection Kit is required for use with MAPmates)
Qty
Catalogue Number
Ordering Description
Qty/Pack
List Price
48-602MAG
Buffer Detection Kit for Magnetic Beads
1 Kit
Space Saver Option Customers purchasing multiple kits may choose to save storage space by eliminating the kit packaging and receiving their multiplex assay components in plastic bags for more compact storage.
This item has been added to favorites.
The Product Has Been Added To Your Cart
You can now customize another kit, choose a premixed kit, check out or close the ordering tool.
FCMAB412F
Sigma-AldrichMilli-Mark® Anti-Collagen Type I-FITC Antibody, clone 5D8-G9
Milli-Mark Anti-Collagen Type I-FITC Antibody, clone 5D8-G9 is an antibody against Collagen Type I for use in FC.
More>>Milli-Mark Anti-Collagen Type I-FITC Antibody, clone 5D8-G9 is an antibody against Collagen Type I for use in FC. Less<<
Milli-Mark® Anti-Collagen Type I-FITC Antibody, clone 5D8-G9 MSDS (material safety data sheet) or SDS, CoA and CoQ, dossiers, brochures and other available documents.
Milli-Mark® Anti-Collagen Type I-FITC Antibody, clone 5D8-G9
Alternate Names
Alpha-1 type I collagen
Background Information
Collagens are highly conserved throughout evolution and are characterised by an uninterrupted "Glycine X Y" triplet repeat that is a necessary part of the triple helical structure. Type I collagen (95 kDa) is found in bone, cornea, skin and tendon. Mutations in the encoding gene are associated with osteogenesis imperfecta, Ehlers Danlos syndrome, and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for Platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor.
References
Product Information
Format
FITC
Control
HEK293T cells
Presentation
Purified mouse monoclonal IgG1 conjugated to FITC in PBS with 0.1% sodium azide and 15 mg/mL BSA.
This gene encodes the pro-alpha1 chains of type I collagen whose triple helix comprises two alpha1 chains and one alpha2 chain. Type I is a fibril-forming collagen found in most connective tissues and is abundant in bone, cornea, dermis and tendon. Mutations in this gene are associated with osteogenesis imperfecta types I-IV, Ehlers-Danlos syndrome type VIIA, Ehlers-Danlos syndrome Classical type, Caffey Disease and idiopathic osteoporosis. Reciprocal translocations between chromosomes 17 and 22, where this gene and the gene for platelet-derived growth factor beta are located, are associated with a particular type of skin tumor called dermatofibrosarcoma protuberans, resulting from unregulated expression of the growth factor. Two transcripts, resulting from the use of alternate polyadenylation signals, have been identified for this gene. [provided by R. Dalgleish]
TISSUE SPECIFICITY: Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite.
PTM: Proline residues at the third position of the tripeptide repeating unit (G-X-Y) are hydroxylated in some or all of the chains. & O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group.
DISEASE: SwissProt: P02452 # Defects in COL1A1 are the cause of Caffey disease [MIM:114000]; also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age. & Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type I (EDS-I) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. Ehlers-Danlos syndrome is a genetically and phenotypically heterogeneous connective-tissue disorder characterized by loose- jointedness and fragile, velvety, stretchable, bruisable skin that heals with peculiar 'cigarette-paper' scars. EDS-I is an autosomal dominant trait. & Defects in COL1A1 are a cause of autosomal dominant Ehlers-Danlos syndrome type VII (EDS-VII) [MIM:130060]; which includes also Ehlers-Danlos syndrome type VII-A1. EDS-VII is characterized by arthrochalasis multiplex congenita, skin hyperextensibility and bruisability. & Defects in COL1A1 are a cause of osteogenesis imperfecta type I (OI-I) [MIM:166200]. OI-I is a dominantly inherited serious newborn disease characterized by bone fragility, normal stature, little or no deformity, blue sclerae and hearing loss in 50% of families. Dentinogenesis imperfecta is rare and may distinguish a subset of OI type I (formation of dentine). & Defects in COL1A1 are a cause of osteogenesis imperfecta type II (OI-II) [MIM:166210]; also known as osteogenesis imperfecta congenita. OI-II is lethal in the perinatal period and is charaterized by calvarial mineralization, beaded ribs, compressed femurs, marked long bone deformity and platyspondyly (congenital flattening of the vertebral bodies). & Defects in COL1A1 are a cause of osteogenesis imperfecta type III (OI-III) [MIM:259420]; also called progressively deforming osteogenesis imperfecta with normal sclerae. OI-III is characterized by progressively deforming bones, usually with moderate deformity at birth, sclerae is variable in color, dentinogenesis imperfecta and hearing loss are common. The stature is very short. & Defects in COL1A1 are a cause of osteogenesis imperfecta type IV (OI-IV) [MIM:166220]. OI-IV is charaterized by normal sclerae, moderate to mild deformity and variable short stature. Dentinogenesis imperfecta is common and hearing loss occurs in some patients. & Genetic variations in COL1A1 are associated with susceptibility to involutional osteoporosis [MIM:166710]; also known as senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mineral density, disrutption of bone microarchitecture, and the alteration of the amount and variety of non-collagenous proteins in bone. Osteoporotic bones are more at risk of fracture. & A chromosomal aberration involving COL1A1 is a cause of dermatofibrosarcoma protuberans (DFSP) [MIM:607907]. Translocation t(17;22)(q22;q13) with PDGF. DFSP is an uncommon, locally aggressive, but rarely metastasizing tumor of the deep dermis and subcutaneous tissue. It typically occurs during early or middle adult life and is most frequently located on the trunk and proximal extremities.
SIMILARITY: SwissProt: P02452 ## Belongs to the fibrillar collagen family. & Contains 1 VWFC domain.
Molecular Weight
139 kDa Calculated
Physicochemical Information
Dimensions
Materials Information
Toxicological Information
Safety Information according to GHS
Safety Information
Product Usage Statements
Quality Assurance
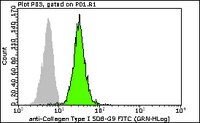
Evaluated by flow cytometry using HEK293T cells.
Usage Statement
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
Storage and Shipping Information
Storage Conditions
Maintain refrigerated at 2-8 °C protected from light in undiluted aliquots for up to 6 months from date of receipt.
Packaging Information
Material Size
100 tests
Transport Information
Supplemental Information
Specifications
Global Trade Item Number
Catalogue Number
GTIN
FCMAB412F
04053252536816
Documentation
Milli-Mark® Anti-Collagen Type I-FITC Antibody, clone 5D8-G9 MSDS