GSK3β inhibition promotes synaptogenesis in Drosophila and mammalian neurons.
Cuesto, G; Jordán-Álvarez, S; Enriquez-Barreto, L; Ferrús, A; Morales, M; Acebes, Á
PloS one
10
e0118475
2015
Show Abstract
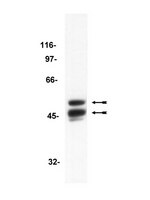
The PI3K-dependent activation of AKT results in the inhibition of GSK3β in most signaling pathways. These kinases regulate multiple neuronal processes including the control of synapse number as shown for Drosophila and rodents. Alzheimer disease's patients exhibit high levels of circulating GSK3β and, consequently, pharmacological strategies based on GSK3β antagonists have been designed. The approach, however, has yielded inconclusive results so far. Here, we carried out a comparative study in Drosophila and rats addressing the role of GSK3β in synaptogenesis. In flies, the genetic inhibition of the shaggy-encoded GSK3β increases the number of synapses, while its upregulation leads to synapse loss. Likewise, in three weeks cultured rat hippocampal neurons, the pharmacological inhibition of GSK3β increases synapse density and Synapsin expression. However, experiments on younger cultures (12 days) yielded an opposite effect, a reduction of synapse density. This unexpected finding seems to unveil an age- and dosage-dependent differential response of mammalian neurons to the stimulation/inhibition of GSK3β, a feature that must be considered in the context of human adult neurogenesis and pharmacological treatments for Alzheimer's disease based on GSK3β antagonists. | Western Blotting | 25764078
 |
Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with β-catenin.
Salazar, VS; Zarkadis, N; Huang, L; Watkins, M; Kading, J; Bonar, S; Norris, J; Mbalaviele, G; Civitelli, R
Journal of cell science
126
5598-609
2013
Show Abstract
Canonical Wnt (cWnt) signaling through β-catenin regulates osteoblast proliferation and differentiation to enhance bone formation. We previously reported that osteogenic action of β-catenin is dependent on BMP signaling. Here, we further examined interactions between cWnt and BMP in bone. In osteoprogenitors stimulated with BMP2, β-catenin localizes to the nucleus, physically interacts with Smad4, and is recruited to DNA-binding transcription complexes containing Smad4, R-Smad1/5 and TCF4. Furthermore, Tcf/Lef-dependent transcription, Ccnd1 expression and proliferation all increase when Smad4, 1 or 5 levels are low, whereas TCF/Lef activities decrease when Smad4 expression is high. The ability of Smad4 to antagonize transcription of Ccnd1 is dependent on DNA-binding activity but Smad4-dependent transcription is not required. In mice, conditional deletion of Smad4 in osterix(+) cells increases mitosis of cells on trabecular bone surfaces as well as in primary osteoblast cultures from adult bone marrow and neonatal calvaria. By contrast, ablation of Smad4 delays differentiation and matrix mineralization by primary osteoblasts in response to Wnt3a, indicating that loss of Smad4 perturbs the balance between proliferation and differentiation in osteoprogenitors. We propose that Smad4 and Tcf/Lef transcription complexes compete for β-catenin, thus restraining cWnt-dependent proliferative signals while favoring the matrix synthesizing activity of osteoblasts. | Western Blotting | 24101723
|
SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties.
Han, K; Holder, JL; Schaaf, CP; Lu, H; Chen, H; Kang, H; Tang, J; Wu, Z; Hao, S; Cheung, SW; Yu, P; Sun, H; Breman, AM; Patel, A; Lu, HC; Zoghbi, HY
Nature
503
72-7
2013
Show Abstract
Mutations in SHANK3 and large duplications of the region spanning SHANK3 both cause a spectrum of neuropsychiatric disorders, indicating that proper SHANK3 dosage is critical for normal brain function. However, SHANK3 overexpression per se has not been established as a cause of human disorders because 22q13 duplications involve several genes. Here we report that Shank3 transgenic mice modelling a human SHANK3 duplication exhibit manic-like behaviour and seizures consistent with synaptic excitatory/inhibitory imbalance. We also identified two patients with hyperkinetic disorders carrying the smallest SHANK3-spanning duplications reported so far. These findings indicate that SHANK3 overexpression causes a hyperkinetic neuropsychiatric disorder. To probe the mechanism underlying the phenotype, we generated a Shank3 in vivo interactome and found that Shank3 directly interacts with the Arp2/3 complex to increase F-actin levels in Shank3 transgenic mice. The mood-stabilizing drug valproate, but not lithium, rescues the manic-like behaviour of Shank3 transgenic mice raising the possibility that this hyperkinetic disorder has a unique pharmacogenetic profile. | Western Blotting | 24153177
|
Chronic renin inhibition with aliskiren improves glucose tolerance, insulin sensitivity, and skeletal muscle glucose transport activity in obese Zucker rats.
Marchionne, EM; Diamond-Stanic, MK; Prasonnarong, M; Henriksen, EJ
American journal of physiology. Regulatory, integrative and comparative physiology
302
R137-42
2012
Show Abstract
We have demonstrated previously that overactivity of the renin-angiotensin system (RAS) is associated with whole body and skeletal muscle insulin resistance in obese Zucker (fa/fa) rats. Moreover, this obesity-associated insulin resistance is reduced by treatment with angiotensin-converting enzyme inhibitors or angiotensin receptor (type 1) blockers. However, it is currently unknown whether specific inhibition of renin itself, the rate-limiting step in RAS functionality, improves insulin action in obesity-associated insulin resistance. Therefore, the present study assessed the effect of chronic, selective renin inhibition using aliskiren on glucose tolerance, whole body insulin sensitivity, and insulin action on the glucose transport system in skeletal muscle of obese Zucker rats. Obese Zucker rats were treated for 21 days with either vehicle or aliskiren (50 mg/kg body wt ip). Renin inhibition was associated with a significant lowering (10%, P less than 0.05) of resting systolic blood pressure and induced reductions in fasting plasma glucose (11%) and free fatty acids (46%) and homeostatic model assessment for insulin resistance (13%). Glucose tolerance (glucose area under the curve) and whole body insulin sensitivity (inverse of the glucose-insulin index) during an oral glucose tolerance test were improved by 15% and 16%, respectively, following chronic renin inhibition. Moreover, insulin-stimulated glucose transport activity in isolated soleus muscle of renin inhibitor-treated animals was increased by 36% and was associated with a 2.2-fold greater Akt Ser(473) phosphorylation. These data provide evidence that chronic selective inhibition of renin activity leads to improvements in glucose tolerance and whole body insulin sensitivity in the insulin-resistant obese Zucker rat. Importantly, chronic renin inhibition is associated with upregulation of insulin action on skeletal muscle glucose transport, and it may involve improved Akt signaling. These data support the strategy of targeting the RAS to improve both blood pressure regulation and insulin action in conditions of insulin resistance. | Western Blotting | 22049232
|
Glycogen synthase kinase 3 regulates PAX3-FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells.
Fu-Yue Zeng,Hanqing Dong,Jimmy Cui,Lingling Liu,Taosheng Chen
Biochemical and biophysical research communications
391
2010
Show Abstract
Patients with alveolar rhabdomyosarcoma (ARMS) have poorer response to conventional chemotherapy and lower survival rates than those with embryonal RMS (ERMS). To identify compounds that preferentially block the growth of ARMS, we conducted a small-scale screen of 160 kinase inhibitors against the ARMS cell line Rh30 and ERMS cell line RD and identified inhibitors of glycogen synthase kinase 3 (GSK3), including TWS119 as ARMS-selective inhibitors. GSK3 inhibitors inhibited cell proliferation and induced apoptosis more effectively in Rh30 than RD cells. Ectopic expression of fusion protein PAX3-FKHR in RD cells significantly increased their sensitivity to TWS119. Down-regulation of GSK3 by GSK3 inhibitors or siRNA significantly reduced the transcriptional activity of PAX3-FKHR. These results suggest that GSK3 is directly involved in regulating the transcriptional activity of PAX3-FKHR. Also, GSK3 phosphorylated PAX3-FKHR in vitro, suggesting that GSK3 might regulate PAX3-FKHR activity via phosphorylation. These findings support a novel mechanism of PAX3-FKHR regulation by GSK3 and provide a novel strategy to develop GSK inhibitors as anti-ARMS therapies. Full Text Article | | 19995556
|
Beta-adrenoceptor stimulation potentiates insulin-stimulated PKB phosphorylation in rat cardiomyocytes via cAMP and PKA.
Stuenaes JT, Bolling A, Ingvaldsen A, Rommundstad C, Sudar E, Lin FC, Lai YC, Jensen J
Br J Pharmacol
160
116-29
2010
Show Abstract
BACKGROUND AND PURPOSE: Genetic approaches have documented protein kinase B (PKB) as a pivotal regulator of heart function. Insulin strongly activates PKB, whereas adrenaline is not considered a major physiological regulator of PKB in heart. In skeletal muscles, however, adrenaline potentiates insulin-stimulated PKB activation without having effect in the absence of insulin. The purpose of the present study was to investigate the interaction between insulin and beta-adrenergic stimulation in regulation of PKB phosphorylation. EXPERIMENTAL APPROACH: Cardiomyocytes were isolated from adult rats by collagenase, and incubated with insulin, isoprenaline, and other compounds. Protein phosphorylation was evaluated by Western blot and phospho-specific antibodies. KEY RESULTS: Isoprenaline increased insulin-stimulated PKB Ser(473) and Thr(308) phosphorylation more than threefold in cardiomyocytes. Isoprenaline alone did not increase PKB phosphorylation. Isoprenaline also increased insulin-stimulated GSK-3beta Ser(9) phosphorylation approximately twofold, supporting that PKB phosphorylation increased kinase activity. Dobutamine (beta(1)-agonist) increased insulin-stimulated PKB phosphorylation as effectively as isoprenaline (more than threefold), whereas salbutamol (beta(2)-agonist) only potentiated insulin-stimulated PKB phosphorylation by approximately 80%. Dobutamine, but not salbutamol, increased phospholamban Ser(16) phosphorylation and glycogen phosphorylase activation (PKA-mediated effects). Furthermore, the cAMP analogue that activates PKA (dibutyryl-cAMP and N(6)-benzoyl-cAMP) increased insulin-stimulated PKB phosphorylation by more than threefold without effect alone. The Epac-specific activator 8-(4-chlorophenylthio)-2'-O-methyl-cAMP (007) increased insulin-stimulated PKB phosphorylation by approximately 50%. Db-cAMP and N(6)-benzoyl-cAMP, but not 007, increased phospholamban Ser(16) phosphorylation. CONCLUSIONS AND IMPLICATIONS: beta-adrenoceptors are strong regulators of PKB phosphorylation via cAMP and PKA when insulin is present. We hypothesize that PKB mediates important signalling in the heart during beta-adrenergic receptors stimulation. | | 20412069
|
Regulation of neural migration by the CREB/CREM transcription factors and altered Dab1 levels in CREB/CREM mutants.
Carmen Díaz-Ruiz,Rosanna Parlato,Fernando Aguado,Jesús M Ureña,Ferran Burgaya,Albert Martínez,Maria A Carmona,Grzegorz Kreiner,Susanne Bleckmann,Jose A Del Río,Günther Schütz,Eduardo Soriano
Molecular and cellular neurosciences
39
2008
Show Abstract
The family of CREB transcription factors is involved in a variety of biological processes including the development and plasticity of the nervous system. To gain further insight into the roles of CREB family members in the development of the embryonic brain, we examined the migratory phenotype of CREB1(Nescre)CREM(-/-) mutants. We found that the lack of CREB/CREM genes is accompanied by anatomical defects in specific layers of the olfactory bulb, hippocampus and cerebral cortex. These changes are associated with decreased Dab1 expression in CREB1(Nescre)CREM(-/-) mutants. Our results indicate that the lack of CREB/CREM genes, specifically in neural and glial progenitors, leads to migration abnormalities during brain development, suggesting that unidentified age-dependent factors modulate the role of CREB/CREM genes in neural development. | | 18786638
|
Divergent cell signaling after short-term intensified endurance training in human skeletal muscle.
Benziane, B; Burton, TJ; Scanlan, B; Galuska, D; Canny, BJ; Chibalin, AV; Zierath, JR; Stepto, NK
American journal of physiology. Endocrinology and metabolism
295
E1427-38
2008
Show Abstract
Endurance training represents one extreme in the continuum of skeletal muscle plasticity. The molecular signals elicited in response to acute and chronic exercise and the integration of multiple intracellular pathways are incompletely understood. We determined the effect of 10 days of intensified cycle training on signal transduction in nine inactive males in response to a 1-h acute bout of cycling at the same absolute workload (164 +/- 9 W). Muscle biopsies were taken at rest and immediately and 3 h after the acute exercise. The metabolic signaling pathways, including AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR), demonstrated divergent regulation by exercise after training. AMPK phosphorylation increased in response to exercise ( approximately 16-fold; P less than 0.05), which was abrogated posttraining (P less than 0.01). In contrast, mTOR phosphorylation increased in response to exercise ( approximately 2-fold; P less than 0.01), which was augmented posttraining (P less than 0.01) in the presence of increased mTOR expression (P less than 0.05). Exercise elicited divergent effects on mitogen-activated protein kinase (MAPK) pathways after training, with exercise-induced extracellular signal-regulated kinase (ERK) 1/2 phosphorylation being abolished (P less than 0.01) and p38 MAPK maintained. Finally, calmodulin kinase II (CaMKII) exercise-induced phosphorylation and activity were maintained (P less than 0.01), despite increased expression ( approximately 2-fold; P less than 0.05). In conclusion, 10 days of intensified endurance training attenuated AMPK, ERK1/2, and mTOR, but not CaMKII and p38 MAPK signaling, highlighting molecular pathways important for rapid functional adaptations and maintenance in response to intensified endurance exercise and training. | | 18827172
|
Akt regulates the survival of vascular smooth muscle cells via inhibition of FoxO3a and GSK3.
Allard, D; Figg, N; Bennett, MR; Littlewood, TD
The Journal of biological chemistry
283
19739-47
2008
Show Abstract
Apoptosis of vascular smooth muscle cells (VSMCs) may lead to atherosclerotic plaque instability and rupture, resulting in myocardial infarction, stroke, and sudden death. However, the molecular mechanisms mediating survival of VSMCs in atherosclerotic plaques remain unknown. Although plaque VSMCs exhibit increased susceptibility to apoptosis and reduced expression of the IGF1 receptor (IGF1R) when compared with normal VSMCs, a causative effect has not been established. Here we show that increased expression of the IGF1R can rescue plaque VSMCs from oxidative stress-induced apoptosis, demonstrating that IGF-1 signaling is a critical regulator of VSMC survival. Akt mediates the majority of the IGF1R survival signaling, and ectopic activation of Akt was sufficient to protect VSMCs in vitro. Both IGF1R and phospho-Akt expression were reduced in human plaque (intimal) VSMCs when compared with medial VSMCs, suggesting that Akt mediates survival signaling in atherosclerosis. Importantly, downstream targets of Akt were identified that mediate its protective effect as inhibition of FoxO3a or GSK3 by Akt-dependent phosphorylation protected VSMCs in vitro. We conclude that Akt and its downstream targets FoxO3a and GSK3 regulate a survival pathway in VSMCs and that their deregulation due to a reduction of IGF1R signaling may promote apoptosis in atherosclerosis. | | 18458087
|
Growth hormone-releasing peptide hexarelin reduces neonatal brain injury and alters Akt/glycogen synthase kinase-3beta phosphorylation.
Brywe, KG; Leverin, AL; Gustavsson, M; Mallard, C; Granata, R; Destefanis, S; Volante, M; Hagberg, H; Ghigo, E; Isgaard, J
Endocrinology
146
4665-72
2005
Show Abstract
Hexarelin (HEX) is a peptide GH secretagogue with a potent ability to stimulate GH secretion and recently reported cardioprotective actions. However, its effects in the brain are largely unknown, and the aim of the present study was to examine the potential protective effect of HEX on the central nervous system after injury, as well as on caspase-3, Akt, and extracellular signal-regulated protein kinase (ERK) signaling cascades in a rat model of neonatal hypoxia-ischemia. Hypoxic-ischemic insult was induced by unilateral carotid ligation and hypoxic exposure (7.7% oxygen), and HEX treatment was administered intracerebroventricularly, directly after the insult. Brain damage was quantified at four coronal levels and by regional neuropathological scoring. Brain damage was reduced by 39% in the treatment group, compared with vehicle group, and injury was significantly reduced in the cerebral cortex, hippocampus, and thalamus but not in the striatum. The cerebroprotective effect was accompanied by a significant reduction of caspase-3 activity and an increased phosphorylation of Akt and glycogen synthase kinase-3beta, whereas ERK was unaffected. In conclusion, we demonstrate for the first time that HEX is neuroprotective in the neonatal setting in vivo and that increased Akt signaling is associated with downstream attenuation of glycogen synthase kinase-3beta activity and caspase-dependent cell death. | | 16081643
|