Persistence of the cell-cycle checkpoint kinase Wee1 in SadA- and SadB-deficient neurons disrupts neuronal polarity.
Myriam Müller,Daniela Lutter,Andreas W Püschel,Myriam Müller,Andreas W Püschel
Journal of cell science
123
2009
Kivonat megmutatása
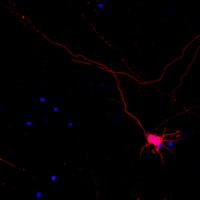
Wee1 is well characterized as a cell-cycle checkpoint kinase that regulates the entry into mitosis in dividing cells. Here we identify a novel function of Wee1 in postmitotic neurons during the establishment of distinct axonal and dendritic compartments, which is an essential step during neuronal development. Wee1 is expressed in unpolarized neurons but is downregulated after neurons have extended an axon. Suppression of Wee1 impairs the formation of minor neurites but does not interfere with axon formation. However, neuronal polarity is disrupted when neurons fail to downregulate Wee1. The kinases SadA and SadB (Sad kinases) phosphorylate Wee1 and are required to initiate its downregulation in polarized neurons. Wee1 expression persists in neurons that are deficient in SadA and SadB and disrupts neuronal polarity. Knockdown of Wee1 rescues the Sada(-/-);Sadb(-/-) mutant phenotype and restores normal polarity in these neurons. Our results demonstrate that the regulation of Wee1 by SadA and SadB kinases is essential for the differentiation of polarized neurons. | 20026642
 |
O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease.
Liu, Fei, et al.
Proc. Natl. Acad. Sci. U.S.A., 101: 10804-9 (2004)
2004
Kivonat megmutatása
Microtubule-associated protein tau is abnormally hyperphosphorylated and aggregated into neurofibrillary tangles in brains of individuals with Alzheimer's disease (AD) and other tauopathies. Tau pathology is critical to pathogenesis and correlates to the severity of dementia. However, the mechanisms leading to abnormal hyperphosphorylation are unknown. Here, we demonstrate that human brain tau was modified by O-GlcNAcylation, a type of protein O-glycosylation by which the monosaccharide beta-N-acetylglucosamine (GlcNAc) attaches to serine/threonine residues via an O-linked glycosidic bond. O-GlcNAcylation regulated phosphorylation of tau in a site-specific manner both in vitro and in vivo. At most of the phosphorylation sites, O-GlcNAcylation negatively regulated tau phosphorylation. In an animal model of starved mice, low glucose uptake/metabolism that mimicked those observed in AD brain produced a decrease in O-GlcNAcylation and consequent hyperphosphorylation of tau at the majority of the phosphorylation sites. The O-GlcNAcylation level in AD brain extracts was decreased as compared to that in controls. These results reveal a mechanism of regulation of tau phosphorylation and suggest that abnormal hyperphosphorylation of tau could result from decreased tau O-GlcNAcylation, which probably is induced by deficient brain glucose uptake/metabolism in AD and other tauopathies. | 15249677
|