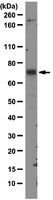
STAT3 regulated ARF expression suppresses prostate cancer metastasis.
Pencik, J; Schlederer, M; Gruber, W; Unger, C; Walker, SM; Chalaris, A; Marié, IJ; Hassler, MR; Javaheri, T; Aksoy, O; Blayney, JK; Prutsch, N; Skucha, A; Herac, M; Krämer, OH; Mazal, P; Grebien, F; Egger, G; Poli, V; Mikulits, W; Eferl, R; Esterbauer, H; Kennedy, R; Fend, F; Scharpf, M; Braun, M; Perner, S; Levy, DE; Malcolm, T; Turner, SD; Haitel, A; Susani, M; Moazzami, A; Rose-John, S; Aberger, F; Merkel, O; Moriggl, R; Culig, Z; Dolznig, H; Kenner, L
Nature communications
6
7736
2015
Kivonat megmutatása
Prostate cancer (PCa) is the most prevalent cancer in men. Hyperactive STAT3 is thought to be oncogenic in PCa. However, targeting of the IL-6/STAT3 axis in PCa patients has failed to provide therapeutic benefit. Here we show that genetic inactivation of Stat3 or IL-6 signalling in a Pten-deficient PCa mouse model accelerates cancer progression leading to metastasis. Mechanistically, we identify p19(ARF) as a direct Stat3 target. Loss of Stat3 signalling disrupts the ARF-Mdm2-p53 tumour suppressor axis bypassing senescence. Strikingly, we also identify STAT3 and CDKN2A mutations in primary human PCa. STAT3 and CDKN2A deletions co-occurred with high frequency in PCa metastases. In accordance, loss of STAT3 and p14(ARF) expression in patient tumours correlates with increased risk of disease recurrence and metastatic PCa. Thus, STAT3 and ARF may be prognostic markers to stratify high from low risk PCa patients. Our findings challenge the current discussion on therapeutic benefit or risk of IL-6/STAT3 inhibition. | 26198641
 |
Transcriptional repressor NIR interacts with the p53-inhibiting ubiquitin ligase MDM2.
Heyne, K; Förster, J; Schüle, R; Roemer, K
Nucleic acids research
42
3565-79
2014
Kivonat megmutatása
NIR (novel INHAT repressor) can bind to p53 at promoters and inhibit p53-mediated gene transactivation by blocking histone acetylation carried out by p300/CBP. Like NIR, the E3 ubiquitin ligase MDM2 can also bind and inhibit p53 at promoters. Here, we present data indicating that NIR, which shuttles between the nucleolus and nucleoplasm, not only binds to p53 but also directly to MDM2, in part via the central acidic and zinc finger domain of MDM2 that is also contacted by several other nucleolus-based MDM2/p53-regulating proteins. Like some of these, NIR was able to inhibit the ubiquitination of MDM2 and stabilize MDM2; however, unlike these nucleolus-based MDM2 regulators, NIR did not inhibit MDM2 to activate p53. Rather, NIR cooperated with MDM2 to repress p53-induced transactivation. This cooperative repression may at least in part involve p300/CBP. We show that NIR can block the acetylation of p53 and MDM2. Non-acetylated p53 has been documented previously to more readily associate with inhibitory MDM2. NIR may thus help to sustain the inhibitory p53:MDM2 complex, and we present evidence suggesting that all three proteins can indeed form a ternary complex. In sum, our findings suggest that NIR can support MDM2 to suppress p53 as a transcriptional activator. | 24413661
|
A novel mechanism of crosstalk between the p53 and NFκB pathways: MDM2 binds and inhibits p65RelA.
Heyne, K; Winter, C; Gerten, F; Schmidt, C; Roemer, K
Cell cycle (Georgetown, Tex.)
12
2479-92
2013
Kivonat megmutatása
The inflammation regulating transcription factor NFκB and the tumor-suppressing transcription factor p53 can act as functional antagonists. Chronic inflammation (NFκB activity) may contribute to the development of cancer through the inhibition of p53 function, while, conversely, p53 activity may dampen inflammation. Here we report that the E3 ubiquitin ligase MDM2, whose gene is transcriptionally activated by both NFκB and p53, can bind and inhibit the p65RelA subunit of NFκB. The interaction is mediated through the N-terminal and the acidic/zinc finger domains of MDM2 on the one hand and through the N-terminal Rel homology domain of p65RelA on the other hand. Co-expression of MDM2 and p65RelA caused ubiquitination of the latter in the nucleus, and this modification was dependent of a functional MDM2 RING domain. Conversely, inhibition of endogenous MDM2 by small-molecule inhibitors or siRNA significantly reduced the ubiquitination of ectopic and endogenous p65RelA. MDM2 was able to equip p65RelA with mutated ubiquitin moieties capable of multiple monoubiquitination but incapable of polyubiquitination; moreover, MDM2 failed to destabilize p65RelA detectably, suggesting that the ubiquitin modification of p65RelA by MDM2 was mostly regulatory rather than stability-determining. MDM2 inhibited the NFκB-mediated transactivation of a reporter gene and the binding of NFκB to its DNA binding motif in vitro. Finally, knockdown of endogenous MDM2 increased the activity of endogenous NFκB as a transactivator. Thus, MDM2 can act as a direct negative regulator of NFκB by binding and inhibiting p65RelA. | 23839035
|