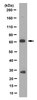
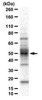
Renal ischemia-reperfusion injury causes intercalated cell-specific disruption of occludin in the collecting duct.
Lee, SY; Shin, JA; Kwon, HM; Weiner, ID; Han, KH
Histochemistry and cell biology
136
637-47
2010
Kivonat megmutatása
Renal ischemic events open tight junctions and disrupt epithelial polarity. The purpose of this study was to examine the effects of ischemia-reperfusion (IR) injury on expression and distribution of the tight junction proteins, occludin and ZO-1, in the rat kidney. IR injury was induced by clamping both renal pedicles for 30 min and animals were killed at 6 h after the reperfusion. IR injury decreased blood bicarbonate level, but did not persistently alter pH, Na(+), K(+), or Cl(-). In control kidneys, occludin immunoreactivity was intense in the tight junctions in the thick ascending limb, distal convoluted tubule, and collecting duct, moderate in the thin limbs of the loop of Henle, and was not detected in the proximal tubule, glomerulus, and blood vessels. ZO-1 was expressed in the same sites in which occludin was expressed, and additionally was also expressed in the proximal tubule, glomerulus, and vascular endothelial cells. IR kidneys exhibited damaged renal tubular epithelial cells in both proximal tubule and collecting duct segments in the outer medulla. In the collecting duct, the response of intercalated cells and principal cells differed. Following IR injury, intercalated cells, but not principal cells, lost their normal epithelial polarity and were frequently extruded into the tubule lumen. Occludin, instead of being localized to tight junctions, was localized diffusely in the cytoplasm in intercalated cells of IR kidneys. Principal cells, in contrast, were not detectably affected and neither occludin nor ZO-1 expression were altered in response to IR injury. The normal localization of ZO-1 expression to tight junction sites in both the proximal tubule and collecting duct was altered in response to IR, and, instead, ZO-1 expression was present diffusely in the cytoplasm. IR injury did not alter detectably either occludin or ZO-1 localization to the tight junction of the thick ascending limb cells. The abundance of total occludin protein by immunoblot analysis was not changed with IR injury. These results demonstrate that renal IR injury causes tight junction disruptions in both the proximal tubule and the collecting duct, and that altered distribution of the tight junction protein, occludin, may play a critical role in the collecting duct dysfunction which IR induces. Teljes cikk | 22048282
 |
Novel roles for metallothionein-I + II (MT-I + II) in defense responses, neurogenesis, and tissue restoration after traumatic brain injury: insights from global gene expression profiling in wild-type and MT-I + II knockout mice.
Milena Penkowa, Mario Cáceres, Rehannah Borup, Finn Cilius Nielsen, Christian Bjørn Poulsen, Albert Quintana, Amalia Molinero, Javier Carrasco, Sergi Florit, Mercedes Giralt, Juan Hidalgo
Journal of neuroscience research
84
1452-74
2005
Kivonat megmutatása
Traumatic injury to the brain is one of the leading causes of injury-related death or disability, especially among young people. Inflammatory processes and oxidative stress likely underlie much of the damage elicited by injury, but the full repertoire of responses involved is not well known. A genomic approach, such as the use of microarrays, provides much insight in this regard, especially if combined with the use of gene-targeted animals. We report here the results of one of these studies comparing wild-type and metallothionein-I + II knockout mice subjected to a cryolesion of the somatosensorial cortex and killed at 0, 1, 4, 8, and 16 days postlesion (dpl) using Affymetrix genechips/oligonucleotide arrays interrogating approximately 10,000 different murine genes (MG_U74Av2). Hierarchical clustering analysis of these genes readily shows an orderly pattern of gene responses at specific times consistent with the processes involved in the initial tissue injury and later regeneration of the parenchyma, as well as a prominent effect of MT-I + II deficiency. The results thoroughly confirmed the importance of the antioxidant proteins MT-I + II in the response of the brain to injury and opened new avenues that were confirmed by immunohistochemistry. Data in KO, MT-I-overexpressing, and MT-II-injected mice strongly suggest a role of these proteins in postlesional activation of neural stem cells. | 16941634
|
Altered expression of aquaporins in bullous keratopathy and Fuchs' dystrophy corneas.
M Cristina Kenney, Shari R Atilano, Nadia Zorapapel, Bret Holguin, Ronald N Gaster, Alexander V Ljubimov
The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society
52
1341-50
2004
Kivonat megmutatása
Corneas with edema-related diseases lose transparency, which causes significant vision loss. This study analyzed seven aquaporins (AQPs) in normal corneas, pseudophakic/aphakic bullous keratopathy (PBK/ABK) corneas, Fuchs' dystrophy corneas, keratoconus corneas, post-cataract surgery (PCS) corneas, and normal organ-cultured corneas. RNA levels for AQP1, AQP4, and beta2-microglobulin were measured by RT-PCR. AQP1 antibody localized to stromal cells of all corneas. PBK/ABK and Fuchs' dystrophy corneas had decreased endothelial cell staining compared with normal. AQP1 mRNA was found in whole corneas and cultured stromal fibroblasts but not in isolated epithelial cells. AQP3 staining was found in basal epithelial cells of the normal, Fuchs' dystrophy, and keratoconus corneas but throughout the entire epithelium of PBK/ABK corneas. AQP4 antibody localized to endothelial cells of all corneas and in stromal cells of PBK/ABK corneas. AQP4 mRNA was identified in whole human corneas. AQP5 was found in epithelial cells of all corneas. AQP0, AQP2, and AQP9 were not found in any corneas. Normal AQP distributions were found in PCS and organ-cultured corneas, although they showed signs of swelling. Our study demonstrates that AQP abnormalities are found in PBK/ABK corneas (decreased AQP1, increased AQP3 and AQP4) and Fuchs' dystrophy corneas (decreased AQP1). Although both have vision-disrupting corneal edema, the mechanisms of fluid accumulation may be different in each disease. | 15385580
|
Renal and blood pressure phenotype in 18-mo-old bradykinin B2R(-/-)CRD mice.
Lisa M Harrison-Bernard, Susana Dipp, Samir S El-Dahr
American journal of physiology. Regulatory, integrative and comparative physiology
285
R782-90
2003
Kivonat megmutatása
Aberrant gene-environment interactions are implicated in the pathogenesis of congenital renal dysgenesis (CRD), a leading cause of renal failure in infants and children. We have recently developed an animal model of CRD that is caused by gestational salt stress (5% NaCl diet; HS) of bradykinin B2R null mice [B2R(-/-)CRD; El-Dahr SS, Harrison-Bernard LM, Dipp S, Yosipiv IV, and Meleg-Smith S. Physiol Genomics 3: 121-131, 2000.]. Developing B2R(-/-)CRD mice exhibit tubular and glomerular cysts, stromal expansion, and loss of corticomedullary differentiation. In addition, B2R(-/-)CRD mice exhibit transient hypertension from 2 to 4 mo of age. The present study was designed to determine the long-term consequences of CRD on renal morphology and salt sensitivity of blood pressure in B2R(-/-)CRD mice. One-year- and 18-mo-old B2R(-/-)CRD mice exhibited stunted renal growth, glomerular cystic abnormalities, and collecting duct ectasia. Moreover, tumors of mesenchymal cell origin emerged in the dysplastic kidneys of 90% of 1-yr-old and 100% of 18-mo-old B2R(-/-)CRD mice but not in age-matched B2R(-/-) or wild-type mice. When challenged with an HS diet, 18-mo-old B2R(-/-)CRD exhibited a significant rise in systolic and diastolic blood pressures and more pronounced natriuresis and diuresis compared with salt-loaded 18-mo-old wild-type mice. Kidney aquaporin-2 expression was decreased by 50%, whereas renin, ANG type 1 receptor, and Na+-K+-ATPase levels were not different in B2R(-/-)CRD mice compared with controls. In conclusion, this study demonstrates that B2R(-/-)CRD mice exhibit permanent phenotypic and functional abnormalities in renal growth and differentiation. This novel model of human disease links gene-environment interactions with renal development and blood pressure homeostasis. | 12805091
|
Water permeability of cochlear outer hair cells: characterization and relationship to electromotility.
Belyantseva, I A, et al.
J. Neurosci., 20: 8996-9003 (2000)
1999
Kivonat megmutatása
The distinguishing feature of the mammalian outer hair cells (OHCs) is to elongate and shorten at acoustic frequencies, when their intracellular potential is changed. This "electromotility" or "electromechanics" depends critically on positive intracellular pressure (turgor), maintained by the inflow of water through yet uncharacterized water pathways. We measured the water volume flow, J(v), across the plasma membrane of isolated guinea pig and rat OHCs after osmotic challenges and estimated the osmotic water permeability coefficient, P(f), to be approximately 10(-2) cm/sec. This value is within the range reported for osmotic flow mediated by the water channel proteins, aquaporins. J(v) was inhibited by HgCl(2), which is known to block aquaporin-mediated water transport. P(f) values that were estimated for OHCs from neonatal rats were of the order of approximately 2 x 10(-3) cm/sec, equivalent to that of membranes lacking water channel proteins. In an immunofluorescence assay we showed that an anti-peptide antibody specific for aquaporins labels the lateral plasma membrane of the OHC in the region in which electromotility is generated. Using patch-clamp recording, we found that water influx into the OHC is regulated by intracellular voltage. We also found that the most pronounced increases of the electromotility-associated charge movement and of the expression of OHC water channels occur between postnatal days 8 and 12, preceding the onset of hearing function in the rat. Our data indicate that electromotility and water transport in OHCs may influence each other structurally and functionally. | 11124975
|
Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing.
Deen, P M, et al.
J. Clin. Invest., 95: 2291-6 (1995)
1994
Kivonat megmutatása
Congenital nephrogenic diabetes insipidus is a recessive hereditary disorder characterized by the inability of the kidney to concentrate urine in response to vasopressin. Recently, we reported mutations in the gene encoding the water channel of the collecting duct, aquaporin-2 (AQP-2) causing an autosomal recessive form of nephrogenic diabetes insipidus (NDI). Expression of these mutant AQP-2 proteins (Gly64Arg, Arg187Cys, Ser216Pro) in Xenopus oocytes revealed nonfunctional water channels. Here we report further studies into the inability of these missense AQP-2 proteins to facilitate water transport in Xenopus oocytes. cRNAs encoding the missense AQPs were translated with equal efficiency as cRNAs encoding wild-type AQP-2 and were equally stable. Arg187Cys AQP2 was more stable and Gly6-4Arg and Ser216Pro AQP2 were less stable when compared to wild-type AQP2 protein. On immunoblots, oocytes expressing missense AQP-2 showed, besides the wild-type 29 kDa band, an endoplasmic reticulum-retarded form of AQP-2 of approximately 32 kD. Immunoblots and immunocytochemistry demonstrated only intense labeling of the plasma membranes of oocytes expressing wild-type AQP-2. Therefore, we conclude that in Xenopus oocytes the inability of Gly64-Arg, Arg187Cys or Ser216Pro substituted AQP-2 proteins to facilitate water transport is caused by an impaired routing to the plasma membrane. | 7537761
|