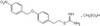
Phytometabolite Dehydroleucodine Induces Cell Cycle Arrest, Apoptosis, and DNA Damage in Human Astrocytoma Cells through p73/p53 Regulation.
Bailon-Moscoso, N; González-Arévalo, G; Velásquez-Rojas, G; Malagon, O; Vidari, G; Zentella-Dehesa, A; Ratovitski, EA; Ostrosky-Wegman, P
PloS one
10
e0136527
2015
显示摘要
Accumulating evidence supports the idea that secondary metabolites obtained from medicinal plants (phytometabolites) may be important contributors in the development of new chemotherapeutic agents to reduce the occurrence or recurrence of cancer. Our study focused on Dehydroleucodine (DhL), a sesquiterpene found in the provinces of Loja and Zamora-Chinchipe. In this study, we showed that DhL displayed cytostatic and cytotoxic activities on the human cerebral astrocytoma D384 cell line. With lactone isolated from Gynoxys verrucosa Wedd, a medicinal plant from Ecuador, we found that DhL induced cell death in D384 cells by triggering cell cycle arrest and inducing apoptosis and DNA damage. We further found that the cell death resulted in the increased expression of CDKN1A and BAX proteins. A marked induction of the levels of total TP73 and phosphorylated TP53, TP73, and γ-H2AX proteins was observed in D384 cells exposed to DhL, but no increase in total TP53 levels was detected. Overall these studies demonstrated the marked effect of DhL on the diminished survival of human astrocytoma cells through the induced expression of TP73 and phosphorylation of TP73 and TP53, suggesting their key roles in the tumor cell response to DhL treatment. | Western Blotting | 26309132
 |
The induction of the p53 tumor suppressor protein bridges the apoptotic and autophagic signaling pathways to regulate cell death in prostate cancer cells.
Ringer, L; Sirajuddin, P; Tricoli, L; Waye, S; Choudhry, MU; Parasido, E; Sivakumar, A; Heckler, M; Naeem, A; Abdelgawad, I; Liu, X; Feldman, AS; Lee, RJ; Wu, CL; Yenugonda, V; Kallakury, B; Dritschilo, A; Lynch, J; Schlegel, R; Rodriguez, O; Pestell, RG; Avantaggiati, ML; Albanese, C
Oncotarget
5
10678-91
2014
显示摘要
The p53 tumor suppressor protein plays a crucial role in influencing cell fate decisions in response to cellular stress. As p53 elicits cell cycle arrest, senescence or apoptosis, the integrity of the p53 pathway is considered a key determinant of anti-tumor responses. p53 can also promote autophagy, however the role of p53-dependent autophagy in chemosensitivity is poorly understood. VMY-1-103 (VMY), a dansylated analog of purvalanol B, displays rapid and potent anti-tumor activities, however the pathways by which VMY works are not fully defined. Using established prostate cancer cell lines and novel conditionally reprogrammed cells (CRCs) derived from prostate cancer patients; we have defined the mechanisms of VMY-induced prostate cancer cell death. Herein, we show that the cytotoxic effects of VMY required a p53-dependent induction of autophagy, and that inhibition of autophagy abrogated VMY-induced cell death. Cancer cell lines harboring p53 missense mutations evaded VMY toxicity and treatment with a small molecule compound that restores p53 activity re-established VMY-induced cell death. The elucidation of the molecular mechanisms governing VMY-dependent cell death in cell lines, and importantly in CRCs, provides the rationale for clinical studies of VMY, alone or in combination with p53 reactivating compounds, in human prostate cancer. | | 25296977
|
Differential effect of hypoxia on etoposide-induced DNA damage response and p53 regulation in different cell types.
Sermeus, Audrey, et al.
J. Cell. Physiol., (2013)
2013
显示摘要
Among the main causes of cancer cell resistance to chemotherapy are p53 mutation and hypoxic tumour microenvironment. However, the effect of hypoxia can be very different from one cell type to the other. We studied the effect of hypoxia on the etoposide-induced cell death in two cancer cell lines, HepG2 and A549 cells. Hypoxia decreased etoposide-induced apoptosis in HepG2 cells but not in A549 cells. Here, we evidenced two pathways, known to play important roles in cancer cell resistance, that are differently affected by hypoxia in these two cell types. First, in HepG2 cells, hypoxia decreased p53 protein level and activity by acting post-transcriptionally and independently of HIF-1. The results suggest an effect of hypoxia on p53 translation. On the other hand, in A549 cells, no effect of hypoxia was observed on p53 level. Secondly, hypoxia decreased DNA damage response in HepG2 cells while this was not the case in A549 cells. Indeed, a decrease in the phosphorylation level of CHK2 and H2AX with a decrease in ATM activity was observed. Importantly, these results evidenced that hypoxia can prevent cancer cell apoptosis by acting at different levels in the cell and that these effects are strongly cell-type dependent. J. Cell. Physiol. © 2013 Wiley Periodicals, Inc. | | 23702906
|
The inhibition of autophagy sensitises colon cancer cells with wild-type p53 but not mutant p53 to topotecan treatment.
Li, DD; Sun, T; Wu, XQ; Chen, SP; Deng, R; Jiang, S; Feng, GK; Pan, JX; Zhang, XS; Zeng, YX; Zhu, XF
PloS one
7
e45058
2012
显示摘要
Topotecan produces DNA damage that induces autophagy in cancer cells. In this study, sensitising topotecan to colon cancer cells with different P53 status via modulation of autophagy was examined.The DNA damage induced by topotecan treatment resulted in cytoprotective autophagy in colon cancer cells with wild-type p53. However, in cells with mutant p53 or p53 knockout, treatment with topotecan induced autophagy-associated cell death. In wild-type p53 colon cancer cells, topotecan treatment activated p53, upregulated the expression of sestrin 2, induced the phosphorylation of the AMPKα subunit at Thr172, and inhibited the mTORC1 pathway. Furthermore, the inhibition of autophagy enhanced the anti-tumour effect of topotecan treatment in wild-type p53 colon cancer cells but alleviated the anti-tumour effect of topotecan treatment in p53 knockout cells in vivo.These results imply that the wild-type p53-dependent induction of cytoprotective autophagy is one of the cellular responses that determines the cellular sensitivity to the DNA-damaging drug topotecan. Therefore, our study provides a potential therapeutic strategy that utilises a combination of DNA-damaging agents and autophagy inhibitors for the treatment of colon cancer with wild-type p53. | Western Blotting | 23024792
|
HDAC5 is required for maintenance of pericentric heterochromatin, and controls cell-cycle progression and survival of human cancer cells.
Peixoto, P, et al.
Cell death and differentiation, (2012)
2012
显示摘要
Histone deacetylases (HDACs) form a family of enzymes, which have fundamental roles in the epigenetic regulation of gene expression and contribute to the growth, differentiation, and apoptosis of cancer cells. In this study, we further investigated the biological function of HDAC5 in cancer cells. We found HDAC5 is associated with actively replicating pericentric heterochromatin during late S phase. We demonstrated that specific depletion of HDAC5 by RNA interference resulted in profound changes in the heterochromatin structure and slowed down ongoing replication forks. This defect in heterochromatin maintenance and assembly are sensed by DNA damage checkpoint pathways, which triggered cancer cells to autophagy and apoptosis, and arrested their growth both in vitro and in vivo. Finally, we also demonstrated that HDAC5 depletion led to enhanced sensitivity of DNA to DNA-damaging agents, suggesting that heterochromatin de-condensation induced by histone HDAC5 silencing may enhance the efficacy of cytotoxic agents that act by targeting DNA in vitro. Together, these results highlighted for the first time an unrecognized link between HDAC5 and the maintenance/assembly of heterochromatin structure, and demonstrated that its specific inhibition might contribute to increase the efficacy of DNA alteration-based cancer therapies in clinic.Cell Death and Differentiation advance online publication, 3 February 2012; doi:10.1038/cdd.2012.3. | | 22301920
|
MG132 inhibition of proteasome blocks apoptosis induced by severe DNA damage.
Zhang, L; Hu, JJ; Gong, F
Cell cycle (Georgetown, Tex.)
10
3515-8
2011
显示摘要
The 26S proteasome, a multicatalytic enzyme complex, is the main intracellular proteolytic system involved in the degradation of ubiquitinated proteins. The ability of proteasome inhibitors to induce apoptosis has been exploited in the recent development of chemotherapeutic agents. Here, we show that inhibition of proteasome by MG132 blocks DNA damage-induced apoptosis. Blockage of apoptosis by MG132 correlates with p53 stabilization and upregulation of p21/WAF1, a p53 transcriptional target. Surprisingly, in the absence of MG132, robust apoptosis induced by a high dose of UV irradiation correlate with rapid p53 degradation. This is in sharp contrast to p53 stabilization when cells were exposed to lower levels of UV irradiation. Our findings highlight a scenario in which severe UV damage can induce rapid p53 degradation by the proteasome. Importantly, these data suggest that the 26S proteasome plays a key role in promoting apoptosis induced by high doses of UV irradiation. | | 22031102
|
Translocation of p53 to mitochondria is regulated by its lipid binding property to anionic phospholipids and it participates in cell death control.
Ching-Hao Li,Yu-Wen Cheng,Po-Ling Liao,Jaw-Jou Kang
Neoplasia (New York, N.Y.)
12
2010
显示摘要
p53, can regulate cell apoptosis in both transcription-dependent and -independent manners. The transcription-independent pathway was demonstrated by the translocation of p53 to mitochondria. Our study showed that p53 mitochondrial translocation was found in mitomycin C (MMC)-treated HepG2. The p53 C-terminal domain is clustered with potential nuclear leading sequences and showed strong electrostatic ion-ion interactions with cardiolipin, phosphatidylglycerol and phosphatidic acid in vitro. Disruption of cardiolipin biosynthesis by phosphatidylglycero-phosphate synthase (PGS) or CDP-diacylglycerol synthase 2 (CDS-2) short hairpin RNA (shRNA) transfection eliminated the MMC-induced translocation of mitochondrial p53. The elimination of mitochondrial p53 translocation also reduced Bcl-xL and Bcl-2 mitochondrial distribution. In HEK 293T models with saturated p53 expression, the mitochondrial partition of p53, Bcl-xL, and Bcl-2 obviously decreased in their PGS shRNA- or CDS-2 shRNA-expressing stable clones. In p53-null H1299 models, both the mitochondrial partitions of Bcl-xL and Bcl-2 were strongly reduced in relation to the HEK 293T models. The Bcl-xL mitochondrial partition was elevated in H1299 models expressing pCEP4-p53wt suggesting the direct carrier role of p53 in transporting Bcl-xL to the mitochondria. We also found that the cytosolic pool of Bcl-xL and Bcl-2 remained unaffected in the low-dose MMC treatment but decreased in the high-dose MMC treatment. The cytosolic pool of Bcl-2 and Bcl-xL directly regulated their amounts in p53-dependent mitochondrial distribution. In the low-dose MMC treatment, the increased mitochondrial p53, Bcl-xL, and Bcl-2 could attenuate apoptosis. However, in the high-dose MMC treatment, only the p53 translocated to the mitochondria and resulted in apoptosis progression. On the basis of this study, we thought mitochondrial p53 might regulate apoptosis in a biphasic manner. 全文本文章 | | 20126473
|
Expression of a homeostatic regulator, Wip1 (wild-type p53-induced phosphatase), is temporally induced by c-Jun and p53 in response to UV irradiation.
Song, JY; Han, HS; Sabapathy, K; Lee, BM; Yu, E; Choi, J
The Journal of biological chemistry
285
9067-76
2010
显示摘要
Wild-type p53-induced phosphatase (Wip1) is induced by p53 in response to stress, which results in the dephosphorylation of proteins (i.e. p38 MAPK, p53, and uracil DNA glycosylase) involved in DNA repair and cell cycle checkpoint pathways. p38 MAPK-p53 signaling is a unique way to induce Wip1 in response to stress. Here, we show that c-Jun directly binds to and activates the Wip1 promoter in response to UV irradiation. The binding of p53 to the promoter occurs earlier than that of c-Jun. In experiments, mutation of the p53 response element (p53RE) or c-Jun consensus sites reduced promoter activity in both non-stressed and stressed A549 cells. Overexpression of p53 significantly decreased Wip1 expression in HCT116 p53(+/+) cells but increased it in HCT116 p53(-/-) cells. Adenovirus-mediated p53 overexpression greatly decreased JNK activity. Up-regulation of Wip1 via the p38 MAPK-p53 and JNK-c-Jun pathways is specific, as demonstrated by our findings that p38 MAPK and JNK inhibitors affected the expression of the Wip1 protein, whereas an ERK inhibitor did not. c-Jun activation occurred much more quickly, and to a greater extent, in A549-E6 cells than in A549 cells, with delayed but fully induced Wip1 expression. These data indicate that Wip1 is activated via both the JNK-c-Jun and p38 MAPK-p53 signaling pathways and that temporal induction of Wip1 depends largely on the balance between c-Jun and p53, which compete for JNK binding. Moreover, our results suggest that JNK-c-Jun-mediated Wip1 induction could serve as a major signaling pathway in human tumors in response to frequent p53 mutation. 全文本文章 | | 20093361
|
The estrogen receptor alpha pathway induces oncogenic Wip1 phosphatase gene expression.
Hye-Sook Han, Eunsil Yu, Ji-Young Song, Ji-Young Park, Se Jin Jang, Jene Choi, Hye-Sook Han, Eunsil Yu, Ji-Young Song, Ji-Young Park, Se Jin Jang, Jene Choi, Hye-Sook Han, Eunsil Yu, Ji-Young Song, Ji-Young Park, Se Jin Jang, Jene Choi
Molecular cancer research : MCR
7
713-23
2009
显示摘要
Wild-type p53-induced phosphatase (Wip1) is a serine/threonine phosphatase induced by DNA-damaging agents. This enzyme dephosphorylates several cell cycle regulating proteins, including p53, p38 mitogen-activated protein kinase, Chk1, and Chk2, resulting in negative feedback regulation of p38-p53 signaling after damage repair. Moreover, the Wip1 gene may be amplified or overexpressed, especially in hormone-regulated organs, and Wip1 gene amplification has been correlated with poor prognosis in hormone-related malignancies, including ovarian cancers. We therefore investigated the link between estrogen signaling and Wip1 expression. We identified seven putative estrogen response elements within 3 kb of the Wip1 promoter. We also found that estradiol (E(2)) treatment produced a 3-fold increase in endogenous Wip1 mRNA and protein expression in MCF7 cells. Direct binding of estrogen receptor (ER)alpha to the Wip1 promoter after E(2) treatment was confirmed by a chromatin immunoprecipitation assay using ERalpha antibody and an electrophoretic mobility shift assay. Wip1 overexpression induced by adenovirus and E(2) facilitated the proliferation of serum-starved ZR-75-1 cells, with cell proliferation induced by overexpressed Wip1 approximately 25% higher than that induced by E(2). Wip1 phosphatase activity was essential for cell cycle progression. Wip1 stimulated the transcriptional activity of its own promoter through E(2)-ERalpha signaling. In addition, Wip1 overexpression induced Rb phosphorylation during cancer cell proliferation. These results indicate that Wip1 up-regulation is important in the pathogenesis of p53(+) and ER(+) breast cancer through the inactivation of p53 by dephosphorylation and the amplification of subsequent estrogenic effects through the E(2)-ERalpha-Wip1 pathway. | | 19435816
|
Hypoxia induces protection against etoposide-induced apoptosis: molecular profiling of changes in gene expression and transcription factor activity.
Sermeus, A; Cosse, JP; Crespin, M; Mainfroid, V; de Longueville, F; Ninane, N; Raes, M; Remacle, J; Michiels, C
Molecular cancer
7
27
2008
显示摘要
it is now well established that hypoxia renders tumor cells resistant to radio- but also chemotherapy. However, few elements are currently available as for the mechanisms underlying this protection.in this study, physiological hypoxia was shown to inhibit apoptosis induced in HepG2 cells by etoposide. Indeed, hypoxia reduced DNA fragmentation, caspase activation and PARP cleavage. The DNA binding activity of 10 transcription factors was followed while the actual transcriptional activity was measured using specific reporter plasmids. Of note is the inhibition of the etoposide-induced activation of p53 under hypoxia. In parallel, data from low density DNA microarrays indicate that the expression of several pro- and anti-apoptotic genes was modified, among which are Bax and Bak whose expression profile paralleled p53 activity. Cluster analysis of data unravels several possible pathways involved in the hypoxia-induced protection against etoposide-induced apoptosis: one of them could be the inhibition of p53 activity under hypoxia since caspase 3 activity parallels Bax and Bak expression profile. Moreover, specific downregulation of HIF-1alpha by RNA interference significantly enhanced apoptosis under hypoxia possibly by preventing the hypoxia mediated decrease in Bak expression without altering Bax expression.these results are a clear demonstration that hypoxia has a direct protective effect on apoptotic cell death. Moreover, molecular profiling points to putative pathways responsible for tumor growth in challenging environmental conditions and cancer cell resistance to chemotherapeutic agents. | | 18366759
|