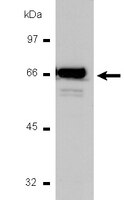
Calcitriol ameliorates renal damage in a pre-established proteinuria model.
Maquigussa, E; Arnoni, CP; Pereira, LG; Boim, MA
Molecular medicine reports
12
1009-15
2015
显示摘要
Proteinuria is critical in the tubulointerstitial changes that ultimately lead to renal insufficiency. Increased protein filtration has direct toxic effects on tubular epithelial cells, leading to epithelial mesenchymal transition (EMT) to a myofibroblast phenotype. Angiotensin II and transforming growth factor (TGF)-β1 are the main mediators of EMT. Calcitriol may exert a potential renoprotective effect by reducing the activity of the renin angiotensin system by suppressing renin gene expression and also by inhibiting the proinflammatory nuclear factor-κB pathway. The present study investigated the benefits of calcitriol treatment in a puromycin-induced proteinuric nephropathy model. Uninephrectomized adult male Wistar rats received intraperitoneal administration of a single dose of puromycin (100 mg/kg) or vehicle. After eight weeks, the animals were divided into two groups and received vehicle or calcitriol (0.5 µg/kg) for four weeks. The vehicle-treated, proteinuric rats developed progressive proteinuria and tubulointerstitial fibrosis after 12 weeks. Increased collagen deposition and fibrosis were significantly ameliorated by calcitriol treatment. Calcitriol was effective in preventing an increase in the EMT markers, α-smooth muscle actin and fibroblast-specific protein 1, reducing macrophage infiltration as evidenced by levels of ED-1. In addition, calcitriol increased the anti-inflammatory cytokine interleukin-10 and reduced the pro-oxidant p47 phox enzyme. These effects were paralleled by a reduction in TGF-β/Smad3 expression. Calcitriol may have therapeutic potential in the proteinuric nephropathy model used in the present study by inhibiting the TGF-β1 axis. | | 25823676
 |
An activated unfolded protein response promotes retinal degeneration and triggers an inflammatory response in the mouse retina.
Rana, T; Shinde, VM; Starr, CR; Kruglov, AA; Boitet, ER; Kotla, P; Zolotukhin, S; Gross, AK; Gorbatyuk, MS
Cell death & disease
5
e1578
2014
显示摘要
Recent studies on the endoplasmic reticulum stress have shown that the unfolded protein response (UPR) is involved in the pathogenesis of inherited retinal degeneration caused by mutant rhodopsin. However, the main question of whether UPR activation actually triggers retinal degeneration remains to be addressed. Thus, in this study, we created a mouse model for retinal degeneration caused by a persistently activated UPR to assess the physiological and morphological parameters associated with this disease state and to highlight a potential mechanism by which the UPR can promote retinal degeneration. We performed an intraocular injection in C57BL6 mice with a known unfolded protein response (UPR) inducer, tunicamycin (Tn) and examined animals by electroretinography (ERG), spectral domain optical coherence tomography (SD-OCT) and histological analyses. We detected a significant loss of photoreceptor function (over 60%) and retinal structure (35%) 30 days post treatment. Analysis of retinal protein extracts demonstrated a significant upregulation of inflammatory markers including interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1) and IBA1. Similarly, we detected a strong inflammatory response in mice expressing either Ter349Glu or T17M rhodopsin (RHO). These mutant rhodopsin species induce severe retinal degeneration and T17M rhodopsin elicits UPR activation when expressed in mice. RNA and protein analysis revealed a significant upregulation of pro- and anti-inflammatory markers such as IL-1β, IL-6, p65 nuclear factor kappa B (NF-kB) and MCP-1, as well as activation of F4/80 and IBA1 microglial markers in both the retinas expressing mutant rhodopsins. We then assessed if the Tn-induced inflammatory marker IL-1β was capable of inducing retinal degeneration by injecting C57BL6 mice with a recombinant IL-1β. We observed ~19% reduction in ERG a-wave amplitudes and a 29% loss of photoreceptor cells compared with control retinas, suggesting a potential link between pro-inflammatory cytokines and retinal pathophysiological effects. Our work demonstrates that in the context of an established animal model for ocular disease, the persistent activation of the UPR could be responsible for promoting retinal degeneration via the UPR-induced pro-inflammatory cytokine IL-1β. | | 25522272
|
miR-146a-5p circuitry uncouples cell proliferation and migration, but not differentiation, in human mesenchymal stem cells.
Hsieh, JY; Huang, TS; Cheng, SM; Lin, WS; Tsai, TN; Lee, OK; Wang, HW
Nucleic acids research
41
9753-63
2013
显示摘要
Administration of mesenchymal stem cells (MSCs) has the potential to ameliorate degenerative disorders and to repair damaged tissues. The homing of transplanted MSCs to injured sites is a critical property of engraftment. Our aim was to identify microRNAs involved in controlling MSC proliferation and migration. MSCs can be isolated from bone marrow and umbilical cord Wharton's jelly (BM-MSCs and WJ-MSCs, respectively), and WJ-MSCs show poorer motility yet have a better amplification rate compared with BM-MSCs. Small RNA sequencing revealed that miR-146a-5p is significantly overexpressed and has high abundance in WJ-MSCs. Knockdown of miR-146a-5p in WJ-MSCs inhibited their proliferation yet enhanced their migration, whereas overexpression of miR-146a-5p in BM-MSCs did not influence their osteogenic and adipogenic potentials. Chemokine (C-X-C motif) ligand 12 (CXCL12), together with SIKE1, which is an I-kappa-B kinase epsilon (IKKε) suppressor, is a direct target of miR-146a-5p in MSCs. Knockdown of miR-146a-5p resulted in the down-regulation of nuclear factor kappa-B (NF-κB) activity, which is highly activated in WJ-MSCs and is known to activate miR-146a-5p promoter. miR-146a-5p is also downstream of CXCL12, and a negative feedback loop is therefore formed in MSCs. These findings suggest that miR-146a-5p is critical to the uncoupling of motility and proliferation of MSCs. Our miRNome data also provide a roadmap for further understanding MSC biology. | Western Blotting | 23963696
|
NF-κB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment.
Huang, G; Wen, Q; Zhao, Y; Gao, Q; Bai, Y
PloS one
8
e61602
2013
显示摘要
CD274, one of two co-stimulatory ligands for programmed death 1 and widely expressed in the mononuclear phagocyte system (MPS), may co-stimulate T cells and regulates inflammatory responses. However, changes in CD274 gene expression and the underlying molecular mechanism are poorly understood during inflammatory responses. Therefore, delineation of the complex mechanisms regulating CD274 expression is critical to understand this immunoregulatory system during inflammatory responses. The purpose of this study was to assess the molecular mechanisms regulating CD274 expression in an in vitro monocyte model of inflammatory response. Firstly, CD274 expression levels in human primary monocytes after lipopolysaccharide (LPS) treatment were observed and correlated with NF-κB activation. Secondly, based on the distribution of putative NF-κB binding sites, 5' truncated human CD274 promoter reporters were constructed, transfected into U937 cells and critical promoter regions for basal (nt -570 to +94) and LPS-induced (nt -1735 to -570) transcription were identified by dual luciferase assays. Finally, a key NF-κB binding site (nt -610 to -601) for LPS-inducible CD274 transcriptional activity was characterized by point mutation analysis and chromatin immunoprecipitation analysis assays (ChIP). Thus, the present study establishes a molecular basis to understand the mechanisms governing CD274 expression in certain infections and inflammatory disorders. | | 23585913
|
Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-κB/HIF-1α signaling pathway.
Kim, HJ; Jeong, JS; Kim, SR; Park, SY; Chae, HJ; Lee, YC
Scientific reports
3
1142
2013
显示摘要
Lipopolysaccharide (LPS) is involved in a variety of inflammatory disorders. Under stress conditions, endoplasmic reticulum (ER) loses the homeostasis in its functions, which is defined as ER stress. Little is known how ER stress is implicated in LPS-induced lung inflammation. In this study, effects of inhibition of ER stress on LPS-induced lung inflammation and transcriptional regulation were examined. An ER stress regulator, 4-phenylbutyrate (PBA) reduced LPS-induced increases of various ER stress markers in the lung. Furthermore, inhibition of ER stress reduced the LPS-induced lung inflammation. Moreover, LPS-induced increases of NF-κB and HIF-1α activity were lowered by inhibition of ER stress. These results suggest that inhibition of ER stress ameliorates LPS-induced lung inflammation through modulation of NF-κB/IκB and HIF-1α signaling pathway. | | 23359618
|
SIMPL enhancement of tumor necrosis factor-α dependent p65-MED1 complex formation is required for mammalian hematopoietic stem and progenitor cell function.
Zhao, W; Breese, E; Bowers, A; Hoggatt, J; Pelus, LM; Broxmeyer, HE; Goebl, M; Harrington, MA
PloS one
8
e61123
2013
显示摘要
Significant insight into the signaling pathways leading to activation of the Rel transcription factor family, collectively termed NF-κB, has been gained. Less well understood is how subsets of NF-κB-dependent genes are regulated in a signal specific manner. The SIMPL protein (signaling molecule that interacts with mouse pelle-like kinase) is required for full Tumor Necrosis Factor-α (TNFα) induced NF-κB activity. We show that SIMPL is required for steady-state hematopoiesis and the expression of a subset of TNFα induced genes whose products regulate hematopoietic cell activity. To gain insight into the mechanism through which SIMPL modulates gene expression we focused on the Tnf gene, an immune response regulator required for steady-state hematopoiesis. In response to TNFα SIMPL localizes to the Tnf gene promoter where it modulates the initiation of Tnf gene transcription. SIMPL binding partners identified by mass spectrometry include proteins involved in transcription and the interaction between SIMPL and MED1 was characterized in more detail. In response to TNFα, SIMPL is found in p65-MED1 complexes where SIMPL enhances p65/MED1/SIMPL complex formation. Together our results indicate that SIMPL functions as a TNFα-dependent p65 co-activator by facilitating the recruitment of MED1 to p65 containing transcriptional complexes to control the expression of a subset of TNFα-induced genes. | | 23630580
|
The response of human macrophages to β-glucans depends on the inflammatory milieu.
Municio, C; Alvarez, Y; Montero, O; Hugo, E; Rodríguez, M; Domingo, E; Alonso, S; Fernández, N; Crespo, MS
PloS one
8
e62016
2013
显示摘要
β-glucans are fungal cell wall components that bind to the C-type lectin-like receptor dectin-1. Polymorphisms of dectin-1 gene are associated with susceptibility to invasive fungal infection and medically refractory ulcerative colitis. The purpose of this study has been addressing the response of human macrophages to β-glucans under different conditions mimicking the composition of the inflammatory milieu in view of the wide plasticity and large range of phenotypical changes showed by these cells, and the relevant role of dectin-1 in several pathophysiological conditions.Serum-differentiated macrophages stimulated with β-glucans showed a low production of TNFα and IL-1β, a high production of IL-6 and IL-23, and a delayed induction of cyclooxygenase-2 and PGE2 biosynthesis that resembled the responses elicited by crystals and those produced when phagosomal degradation of the phagocytic cargo increases ligand access to intracellular pattern recognition receptors. Priming with a low concentration of LPS produced a rapid induction of cyclooxygenase-2 and a synergistic release of PGE2. When the differentiation of the macrophages was carried out in the presence of M-CSF, an increased expression of dectin-1 B isoform was observed. In addition, this treatment made the cells capable to release arachidonic acid in response to β-glucan.These results indicate that the macrophage response to fungal β-glucans is strongly influenced by cytokines and microbial-derived factors that are usual components of the inflammatory milieu. These responses can be sorted into three main patterns i) an elementary response dependent on phagosomal processing of pathogen-associated molecular patterns and/or receptor-independent, direct membrane binding linked to the immunoreceptor tyrosine-based activation motif-bearing transmembrane adaptor DNAX-activating protein 12, ii) a response primed by TLR4-dependent signals, and iii) a response dependent on M-CSF and dectin-1 B isoform expression that mainly signals through the dectin-1 B/spleen tyrosine kinase/cytosolic phospholipase A2 route. | Western Blotting | 23637950
|
BCL-3 attenuation of TNFA expression involves an incoherent feed-forward loop regulated by chromatin structure.
Walker, T; Adamson, A; Jackson, DA
PloS one
8
e77015
2013
显示摘要
Induction of genes is rarely an isolated event; more typically occurring as part of a web of parallel interactions, or motifs, which act to refine and control gene expression. Here, we define an Incoherent Feed-forward Loop motif in which TNFα-induced NF-κB signalling activates expression of the TNFA gene itself and also controls synthesis of the negative regulator BCL-3. While sharing a common inductive signal, the two genes have distinct temporal expression profiles. Notably, while the TNFA gene promoter is primed to respond immediately to activated NF-κB in the nucleus, induction of BCL3 expression only occurs after a time delay of about 1h. We show that this time delay is defined by remodelling of the BCL3 gene promoter, which is required to activate gene expression, and characterise the chromatin delayed induction of BCL3 expression using mathematical models. The models show how a delay in inhibitor production effectively uncouples the rate of response to inflammatory cues from the final magnitude of inhibition. Hence, within this regulatory motif, a delayed (incoherent) feed-forward loop together with differential rates of TNFA (fast) and BCL3 (slow) mRNA turnover provide robust, pulsatile expression of TNFα . We propose that the structure of the BCL-3-dependent regulatory motif has a beneficial role in modulating expression dynamics and the inflammatory response while minimising the risk of pathological hyper-inflammation. | | 24130828
|
Glutamine modulates lipopolysaccharide-induced activation of NF-κB via the Akt/mTOR pathway in lung epithelial cells.
Hou, YC; Chiu, WC; Yeh, CL; Yeh, SL
American journal of physiology. Lung cellular and molecular physiology
302
L174-83
2012
显示摘要
Lung epithelial cells are important barriers in the respiratory system that provoke inflammatory responses through nuclear factor (NF)-κB activation to prevent pathogens from invading the body. Lipopolysaccharide (LPS) is a common pathogen-associated stimulus that activates IκB kinase (IKK) to regulate NF-κB-mediated inflammation through modulating nuclear translocation and phosphorylation of NF-κB. Previously, it was shown that Akt and the mammalian target of rapamycin (mTOR) are involved in the phosphorylation of IKK to activate NF-κB. Herein, we demonstrate that glutamine (GLN) modulated LPS-induced activation of NF-κB through the Akt/mTOR/IKK pathway in BEAS-2B cells. BEAS-2B cells in submerged culture were placed in medium containing different concentrations of GLN (0, 0.5, 1, and 2.5 mM) with 1 μg/ml LPS. Results showed that GLN deprivation induced phosphorylation of Akt/mTOR/IKK signaling, increased levels of NF-κB nuclear translocation and phosphorylated NF-κB, and upregulated NF-κB-dependent transcriptional activity, which was suppressed by GLN administration. Expressions of NF-κB-targeted genes were also reduced by supplemental GLN. GLN administration improved cell viability, whereas 0.5 mM GLN had a greater extent of inhibition on the Akt/mTOR/IKK/NF-κB signaling cascade. The inhibitory effects of GLN on NF-κB activation were also observed in cells cultured under air-liquid interface condition. These results indicate that GLN deprivation increased LPS-induced NF-κB activation and transcriptional activity, which was reversed by GLN administration. The findings provide potential mechanisms of GLN's modulation of LPS-induced NF-κB activation in lung epithelial cells and imply that maintaining a physiological concentration of GLN is essential in preventing LPS-induced lung inflammation. | | 22003094
|
IKKα and alternative NF-κB regulate PGC-1β to promote oxidative muscle metabolism.
Bakkar, N; Ladner, K; Canan, BD; Liyanarachchi, S; Bal, NC; Pant, M; Periasamy, M; Li, Q; Janssen, PM; Guttridge, DC
The Journal of cell biology
196
497-511
2012
显示摘要
Although the physiological basis of canonical or classical IκB kinase β (IKKβ)-nuclear factor κB (NF-κB) signaling pathway is well established, how alternative NF-κB signaling functions beyond its role in lymphoid development remains unclear. In particular, alternative NF-κB signaling has been linked with cellular metabolism, but this relationship is poorly understood. In this study, we show that mice deleted for the alternative NF-κB components IKKα or RelB have reduced mitochondrial content and function. Conversely, expressing alternative, but not classical, NF-κB pathway components in skeletal muscle stimulates mitochondrial biogenesis and specifies slow twitch fibers, suggesting that oxidative metabolism in muscle is selectively controlled by the alternative pathway. The alternative NF-κB pathway mediates this specificity by direct transcriptional activation of the mitochondrial regulator PPAR-γ coactivator 1β (PGC-1β) but not PGC-1α. Regulation of PGC-1β by IKKα/RelB also is mammalian target of rapamycin (mTOR) dependent, highlighting a cross talk between mTOR and NF-κB in muscle metabolism. Together, these data provide insight on PGC-1β regulation during skeletal myogenesis and reveal a unique function of alternative NF-κB signaling in promoting an oxidative metabolic phenotype. | | 22351927
|