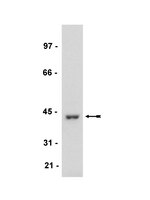
MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition.
Taylor, DM; Moser, R; Régulier, E; Breuillaud, L; Dixon, M; Beesen, AA; Elliston, L; Silva Santos, Mde F; Kim, J; Jones, L; Goldstein, DR; Ferrante, RJ; Luthi-Carter, R
The Journal of neuroscience : the official journal of the Society for Neuroscience
33
2313-25
2013
显示摘要
We previously demonstrated that sodium butyrate is neuroprotective in Huntington's disease (HD) mice and that this therapeutic effect is associated with increased expression of mitogen-activated protein kinase/dual-specificity phosphatase 1 (MKP-1/DUSP1). Here we show that enhancing MKP-1 expression is sufficient to achieve neuroprotection in lentiviral models of HD. Wild-type MKP-1 overexpression inhibited apoptosis in primary striatal neurons exposed to an N-terminal fragment of polyglutamine-expanded huntingtin (Htt171-82Q), blocking caspase-3 activation and significantly reducing neuronal cell death. This neuroprotective effect of MKP-1 was demonstrated to be dependent on its enzymatic activity, being ablated by mutation of its phosphatase domain and being attributed to inhibition of specific MAP kinases (MAPKs). Overexpression of MKP-1 prevented the polyglutamine-expanded huntingtin-induced activation of c-Jun N-terminal kinases (JNKs) and p38 MAPKs, whereas extracellular signal-regulated kinase (ERK) 1/2 activation was not altered by either polyglutamine-expanded Htt or MKP-1. Moreover, mutants of MKP-1 that selectively prevented p38 or JNK binding confirmed the important dual contributions of p38 and JNK regulation to MKP-1-mediated neuroprotection. These results demonstrate additive effects of p38 and JNK MAPK inhibition by MKP-1 without consequence to ERK activation in this striatal neuron-based paradigm. MKP-1 also provided neuroprotection in vivo in a lentiviral model of HD neuropathology in rat striatum. Together, these data extend previous evidence that JNK- and p38-mediated pathways contribute to HD pathogenesis and, importantly, show that therapies simultaneously inhibiting both JNK and p38 signaling pathways may lead to improved neuroprotective outcomes. | Western Blotting, Immunohistochemistry, Immunocytochemistry | 23392662
 |
Epinephrine and AICAR-induced PGC-1α mRNA expression is intact in skeletal muscle from rats fed a high-fat diet.
Frier, BC; Wan, Z; Williams, DB; Stefanson, AL; Wright, DC
American journal of physiology. Cell physiology
302
C1772-9
2012
显示摘要
Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a master regulator of mitochondrial biogenesis and is controlled, at least in part, through AMP-activated protein kinase and p38-dependent pathways. There is evidence demonstrating that activation of these kinases and induction of PGC-1α in skeletal muscle are regulated by catecholamines. The purpose of the present study was to determine if consumption of a high-fat diet (HFD) impairs epinephrine and 5-aminoimidazole-4-carboxamide-1β-d-ribofuranoside (AICAR) signaling and induction of PGC-1α in rat skeletal muscle. Male Wistar rats were fed chow or a HFD for 6 wk and then given a weight-adjusted bolus injection of epinephrine (20, 10, or 5 μg/100 g body wt sc) or saline, and triceps muscles were harvested 30 min (signaling) or 2 and 4 h (gene expression) postinjection. Despite blunted increases in p38 phosphorylation, the ability of epinephrine to induce PGC-1α was intact in skeletal muscle from HFD-fed rats and was associated with normal increases in activation of PKA and phosphorylation of cAMP response element-binding protein, reputed mediators of PGC-1α expression. The attenuated epinephrine-mediated increase in p38 phosphorylation was independent of increases in MAPK phosphatase 1. At 2 h following AICAR treatment (0.5 g/kg body wt sc), AMP-activated protein kinase and acetyl-CoA carboxylase phosphorylation were similar in skeletal muscle from chow- and HFD-fed rats. Surprisingly, AICAR-induced increases in PGC-1α mRNA levels were greater in skeletal muscle from HFD-fed rats. Our results demonstrate that the ability of epinephrine and AICAR to induce PGC-1α remains intact in skeletal muscle from HFD-fed rats. These results question the existence of reduced β-adrenergic responsiveness in diet-induced obesity and demonstrate that increases in p38 phosphorylation are not required for induction of PGC-1α in muscle from obese rats. | | 22496244
|
A genome-wide RNAi screen reveals MAP kinase phosphatases as key ERK pathway regulators during embryonic stem cell differentiation.
Yang, SH; Kalkan, T; Morrisroe, C; Smith, A; Sharrocks, AD
PLoS genetics
8
e1003112
2012
显示摘要
Embryonic stem cells and induced pluripotent stem cells represent potentially important therapeutic agents in regenerative medicine. Complex interlinked transcriptional and signaling networks control the fate of these cells towards maintenance of pluripotency or differentiation. In this study we have focused on how mouse embryonic stem cells begin to differentiate and lose pluripotency and, in particular, the role that the ERK MAP kinase and GSK3 signaling pathways play in this process. Through a genome-wide siRNA screen we have identified more than 400 genes involved in loss of pluripotency and promoting the onset of differentiation. These genes were functionally associated with the ERK and/or GSK3 pathways, providing an important resource for studying the roles of these pathways in controlling escape from the pluripotent ground state. More detailed analysis identified MAP kinase phosphatases as a focal point of regulation and demonstrated an important role for these enzymes in controlling ERK activation kinetics and subsequently determining early embryonic stem cell fate decisions. | Western Blotting | 23271975
|
Modulation of gonadotropin-releasing hormone-induced extracellular signal-regulated kinase activation by dual-specificity protein phosphatase 1 in LbetaT2 gonadotropes.
Nguyen, KA; Intriago, RE; Upadhyay, HC; Santos, SJ; Webster, NJ; Lawson, MA
Endocrinology
151
4882-93
2010
显示摘要
As the regulator of pituitary reproductive hormone synthesis, the hypothalamic neuropeptide GnRH is the central regulator of reproduction. A hallmark of GnRH action is the differential control of gene expression in pituitary gonadotropes through varied pulsatile stimulation. Among other signaling events, GnRH activation of the ERK family of MAPKs plays a significant role in the transcriptional regulation of the luteinizing hormone β-subunit gene and regulation of cap-dependent translation. We evaluated the ERK response to different GnRH pulse amplitudes in the gonadotrope cell line LβT2. We found that low-amplitude stimulation with GnRH invokes a rapid and transient ERK activation, whereas high-amplitude stimulation invokes a prolonged activation specifically in the cytoplasm fraction of LβT2 cells. Nuclear and cytoplasmic targets of ERK, Ets-like gene 1, and eukaryotic initiation factor 4E, respectively, are similarly activated. Feedback control of ERK activation occurs mainly through the dual-specificity protein phosphatases (DUSPs). DUSP1 is localized to the nucleus in LβT2 cells but DUSP4, another member implicated in GnRH feedback, exists in both the nucleus and cytoplasm. Manipulation of nuclear DUSP activity through overexpression or knockdown of Dusp1 modulates the ERK response to low and high GnRH pulse amplitudes and activation of the Lhb promoter. Dusp1 overexpression abolishes sustained ERK activation and inhibits Lhb promoter activity induced by high amplitude pulses. Conversely, Dusp1 knockdown enhances ERK activation by low-amplitude stimulation and increases stimulation of Lhb promoter activity. We conclude that DUSP1 feedback activity modulates ERK activation and the transcriptional response to GnRH. 全文本文章 | | 20685880
|
Role of MAPK phosphatase-1 in sustained activation of JNK during ethanol-induced apoptosis in hepatocyte-like VL-17A cells.
Venugopal, SK; Chen, J; Zhang, Y; Clemens, D; Follenzi, A; Zern, MA
The Journal of biological chemistry
282
31900-8
2007
显示摘要
Ethanol metabolism plays a central role in activating the mitogen-activated protein kinase (MAPK) cascade leading to inflammation and apoptosis. Sustained activation of c-Jun N-terminal kinase (JNK), one of the MAPKs, has been shown to induce apoptosis in hepatocytes. MAPK phosphatase-1 (MKP-1) has been shown to dephosphorylate MAPKs in several cells. The aim of the study is to evaluate the role of MKP-1 in sustained JNK activation as a mechanism to explain ethanol-induced hepatocyte apoptosis. VL-17A cells (HepG2 cells overexpressing alcohol dehydrogenase and cytochrome P450-2E1) were exposed to ethanol for different time periods. Western blots were performed for MKP-1, phospho-JNK, phosphotyrosine, and protein kinase Cdelta (PKCdelta). Electrophoretic mobility shift assays for AP-1 were performed. Apoptosis was measured by caspase-3 activity assay, TUNEL, and 4',6-diamidino-2-phenylindole staining. Reactive oxygen species were neutralized by overexpressing both superoxide dismutase-3 and catalase genes using lentiviral vectors in VL-17A cells. Ethanol incubation markedly decreased the MKP-1 protein levels to 15% of control levels and was associated with sustained phosphorylation of p46 JNK and p54 JNK, as well as increased apoptosis. VL-17A cells overexpressing superoxide dismutase-3 and catalase, treatment with a tyrosine kinase inhibitor, or incubation of the cells with PKCdelta small interference RNAs significantly inhibited the ethanol-induced MKP-1 degradation and apoptosis. Ethanol-induced oxidative stress enhanced the tyrosine phosphorylation of PKCdelta, which in turn caused the proteasomal degradation of MKP-1, leading to sustained JNK activation and increased apoptosis in VL-17A cells. | Western Blotting | 17848570
|
Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of toll-like receptor 2.
Imasato, Akira, et al.
J. Biol. Chem., 277: 47444-50 (2002)
2002
| | 12356755
|
Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38.
Lasa, Marina, et al.
Mol. Cell. Biol., 22: 7802-11 (2002)
2002
显示摘要
The stress-activated protein kinase p38 stabilizes a number of mRNAs encoding inflammatory mediators, such as cyclooxygenase 2 (Cox-2). In HeLa cells the anti-inflammatory glucocorticoid dexamethasone destabilizes Cox-2 mRNA by inhibiting p38 function. Here we demonstrate that this effect is phosphatase dependent. Furthermore, in HeLa cells dexamethasone induced the sustained expression of mitogen-activated protein kinase phosphatase 1 (MKP-1), a potent inhibitor of p38 function. The inhibition of p38 and the induction of MKP-1 by dexamethasone occurred with similar dose dependence and kinetics. No other known p38 phosphatases were induced by dexamethasone, and other cell types which failed to express MKP-1 also failed to inhibit p38 in response to dexamethasone. The proinflammatory cytokine interleukin 1 (IL-1) induced MKP-1 expression in a p38-dependent manner and acted synergistically with dexamethasone to induce MKP-1 expression. In HeLa cells treated with IL-1 or IL-1 and dexamethasone, the dynamics of p38 activation mirrored the expression of MKP-1. These observations suggest that MKP-1 participates in a negative-feedback loop which regulates p38 function and that dexamethasone may inhibit proinflammatory gene expression in part by inducing MKP-1 expression. | | 12391149
|
Dephosphorylation and inactivation of the mitogen-activated protein kinase by a mitogen-induced Thr/Tyr protein phosphatase.
Zheng, C F and Guan, K L
J. Biol. Chem., 268: 16116-9 (1993)
1993
显示摘要
The activation of extracellular signal-regulated kinase (ERK) or mitogen-activated protein kinase (MAPK) by a dual specific kinase, MEK (MAPK or ERK kinase), is a critical event in the mitogenic signal transduction pathway. However, little is known about the mechanism of ERK inactivation, which occurs after stimulation. In this report, we demonstrated that a dual specific protein phosphatase, HVH1 (human VH1 phosphatase homolog) whose expression is induced by mitogenic growth factors, specifically inactivates ERK. When several phosphoproteins were tested for recombinant HVH1, only MEK-activated ERK1 was dephosphorylated. HVH1 selectively dephosphorylated threonine and tyrosine residues but not serine residues of the activated ERK1. Inactivation of ERK1 by HVH1 could be reversed by MEK, suggesting that HVH1 dephosphorylates the same residues that are recognized and phosphorylated by MEK. Our results suggest that mitogenic growth factors transiently activate ERK (peak at 5 min followed by a rapid decline) by temporally activating MEK (the on signal) and inducing the expression of HVH phosphatase (the off signal). | | 8344896
|