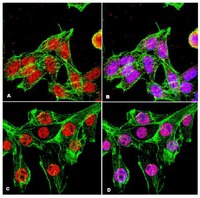
Loss of INPP4B causes a DNA repair defect through loss of BRCA1, ATM and ATR and can be targeted with PARP inhibitor treatment.
Ip, LR; Poulogiannis, G; Viciano, FC; Sasaki, J; Kofuji, S; Spanswick, VJ; Hochhauser, D; Hartley, JA; Sasaki, T; Gewinner, CA
Oncotarget
6
10548-62
2015
显示摘要
Treatment options for ovarian cancer patients remain limited and overall survival is less than 50% despite recent clinical advances. The lipid phosphatase inositol polyphosphate 4-phosphatase type II (INPP4B) has been described as a tumor suppressor in the PI3K/Akt pathway with loss of expression found most pronounced in breast, ovarian cancer and melanoma. Using microarray technology we identified a DNA repair defect in INPP4B-deficient cells, which we further characterized by comet assays and quantification of γH2AX, RAD51 and 53BP1 foci formation. INPP4B loss resulted in significantly increased sensitivity towards PARP inhibition, comparable to loss of BRCA1 in two- and three-dimensional in vitro models, as well as in in vivo xenograft models. Mechanistically, we discovered that INPP4B forms a protein complex with the key players of DNA repair, ATR and BRCA1, in GST pulldown and 293T overexpression assays, and INPP4B loss affects BRCA1, ATM and ATR protein stability resulting in the observed DNA repair defect. Given that INPP4B loss has been found in 40% of ovarian cancer patients, this study provides the rationale for establishing INPP4B as a biomarker of PARP inhibitor response, and consequently offers novel therapeutic options for a significant subset of patients. Loss of the tumor suppressor inositol polyphosphate 4-phosphatase type II (INPP4B) results in a DNA repair defect due to concomitant loss of BRCA1, ATR and ATM and can be therapeutically targeted with PARP inhibitors. | Western Blotting | 25868852
 |
Characterization of LGALS3 (galectin-3) as a player in DNA damage response.
Carvalho, RS; Fernandes, VC; Nepomuceno, TC; Rodrigues, DC; Woods, NT; Suarez-Kurtz, G; Chammas, R; Monteiro, AN; Carvalho, MA
Cancer biology & therapy
15
840-50
2014
显示摘要
DNA damage repair (DDR) is an orchestrated process encompassing the injury detection to its complete resolution. DNA double-strand break lesions are repaired mainly by two distinct mechanisms: the error-free homologous recombination (HR) and the error-prone non-homologous end-joining. Galectin-3 (GAL3) is the unique member of the chimeric galectins subfamily and is reported to be involved in several cancer development and progression related events. Recently our group described a putative protein interaction between GAL3 and BARD1, the main partner of breast and ovarian cancer susceptibility gene product BRCA1, both involved in HR pathway. In this report we characterized GAL3/BARD1 protein interaction and evaluated the role of GAL3 in DDR pathways using GAL3 silenced human cells exposed to different DNA damage agents. In the absence of GAL3 we observed a delayed DDR response activation, as well as a decrease in the G 2/M cell cycle checkpoint arrest associated with HR pathway. Moreover, using a TAP-MS approach we also determined the protein interaction network of GAL3. | | 24755837
|
Wild-type p53-induced phosphatase 1 (Wip1) forestalls cellular premature senescence at physiological oxygen levels by regulating DNA damage response signaling during DNA replication.
Sakai, H; Fujigaki, H; Mazur, SJ; Appella, E
Cell cycle (Georgetown, Tex.)
13
1015-29
2014
显示摘要
Wip1 (protein phosphatase Mg(2+)/Mn(2+)-dependent 1D, Ppm1d) is a nuclear serine/threonine protein phosphatase that is induced by p53 following the activation of DNA damage response (DDR) signaling. Ppm1d(-/-) mouse embryonic fibroblasts (MEFs) exhibit premature senescence under conventional culture conditions; however, little is known regarding the role of Wip1 in regulating cellular senescence. In this study, we found that even at a representative physiological concentration of 3% O2, Ppm1d(-/-) MEFs underwent premature cellular senescence that depended on the functional activation of p53. Interestingly, Ppm1d(-/-) MEFs showed increased H2AX phosphorylation levels without increased levels of reactive oxygen species (ROS) or DNA base damage compared with wild-type (Wt) MEFs, suggesting a decreased threshold for DDR activation or sustained DDR activation during recovery. Notably, the increased H2AX phosphorylation levels observed in Ppm1d(-/-) MEFs were primarily associated with S-phase cells and predominantly dependent on the activation of ATM. Moreover, these same phenotypes were observed when Wt and Ppm1d(-/-) MEFs were either transiently or chronically exposed to low levels of agents that induce replication-mediated double-stranded breaks. These findings suggest that Wip1 prevents the induction of cellular senescence at physiological oxygen levels by attenuating DDR signaling in response to endogenous double-stranded breaks that form during DNA replication. | Immunocytochemistry | 24552809
|
Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase.
Jin, J; Ang, XL; Ye, X; Livingstone, M; Harper, JW
The Journal of biological chemistry
283
19322-8
2008
显示摘要
In response to DNA damage, cells activate a signaling pathway that promotes cell cycle arrest and degradation of the cell cycle regulator Cdc25A. Cdc25A degradation occurs via the SCFbeta-TRCP pathway and phosphorylation of Ser-76. Previous work indicates that the checkpoint kinase Checkpoint kinase 1 (Chk1) is capable of phosphorylating Ser-76 in Cdc25A, thereby promoting its degradation. In contrast, other experiments involving overexpression of dominant Chk2 mutant proteins point to a role for Chk2 in Cdc25A degradation. However, loss-of-function studies that implicate Chk2 in Cdc25A turnover are lacking, and there is no evidence that Chk2 is capable of phosphorylating Ser-76 in Cdc25A despite the finding that Chk1 and Chk2 sometimes share overlapping primary specificity. We find that although Chk2 can phosphorylate many of the same sites in Cdc25A that Chk1 phosphorylates, albeit with reduced efficiency, Chk2 is unable to efficiently phosphorylate Ser-76. Consistent with this, Chk2, unlike Chk1, is unable to support SCFbeta-TRCP-mediated ubiquitination of Cdc25A in vitro. In CHK2(-/-) HCT116 cells, the kinetics of Cdc25A degradation in response to ionizing radiation is comparable with that seen in HCT116 cells containing Chk2, indicating that Chk2 is not generally required for timely DNA damage-dependent Cdc25A turnover. In contrast, depletion of Chk1 by RNA interference in CHK2(-/-) cells leads to Cdc25A stabilization in response to ionizing radiation. These data support the idea that Chk1 is the primary signal transducer linking activation of the ATM/ATR kinases to Cdc25A destruction in response to ionizing radiation. | | 18480045
|
Regulation of intra-S phase checkpoint by ionizing radiation (IR)-dependent and IR-independent phosphorylation of SMC3.
Luo, H; Li, Y; Mu, JJ; Zhang, J; Tonaka, T; Hamamori, Y; Jung, SY; Wang, Y; Qin, J
The Journal of biological chemistry
283
19176-83
2008
显示摘要
Structure maintenance of chromosome 1 (SMC1) is phosphorylated by ataxia telangiectasia-mutated (ATM) in response to ionizing radiation (IR) to activate intra-S phase checkpoint. A role of CK2 in DNA damage response has been implicated in many previous works, but the molecular mechanism for its activation is not clear. In the present work, we report that SMC3 is phosphorylated at Ser-1067 and Ser-1083 in vivo. Ser-1083 phosphorylation is IR-inducible, depends on ATM and Nijmegen breakage syndrome 1 (NBS1), and is required for intra-S phase checkpoint. Interestingly, Ser-1067 phosphorylation is constitutive and is not induced by IR but also affects intra-S phase checkpoint. Phosphorylation of Ser-1083 is weakened in cells expressing S1067A mutant, suggesting interplay between Ser-1067 and Ser-1083 phosphorylation in DNA damage response. Consistently, small interfering RNA knockdown of CK2 leads to attenuated phosphorylation of Ser-1067 as well as intra-S phase checkpoint defect. Our data provide evidence that phosphorylation of a core cohesin subunit SMC3 by ATM plays an important role in DNA damage response and suggest that a constitutive phosphorylation by CK2 may affect intra-S phase checkpoint by modulating SMC3 phosphorylation by ATM. | | 18442975
|
Delayed mammary gland involution in MMTV-AKT1 transgenic mice.
Ackler, S; Ahmad, S; Tobias, C; Johnson, MD; Glazer, RI
Oncogene
21
198-206
2002
显示摘要
AKT1/protein kinase Balpha is a protein-serine/threonine kinase that regulates multiple targets involved in cell survival and cell cycle progression in a variety of cell types including breast cancer cells. To explore the role of Akt1 in mammary gland function and tumorigenesis, transgenic mice were generated that express human AKT1 under the control of the MMTV promoter. Virgin transgenic mice did not exhibit a dominant phenotype, but upon cessation of lactation, a notable delay in involution occurred compared to age-matched non-transgenic mice. The delay in involution coincided with increased hyperplasia as evidenced by an increased number of binucleated epithelial cells and a marked elevation in cyclin D1 expression in mammary epithelium. The delayed involution phenotype corresponded to increased phosphorylation of Thr308 in AKT1 and Ser136 in BAD, but not phosphorylation of Ser21 in GSK-3alpha. There was no evidence of mammary dysplasia or neoplasia during the lifespan of multiparous transgenic mice. These data suggest that AKT1 is involved in cell survival in the lactating and involuting mammary gland, but that overexpression of AKT1 alone is insufficient to induce transformation. | | 11803463
|
Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation.
James, SR; Downes, CP; Gigg, R; Grove, SJ; Holmes, AB; Alessi, DR
The Biochemical journal
315 ( Pt 3)
709-13
1996
显示摘要
Recent evidence has suggested that activation of phosphoinositide 3-kinase (PI 3-kinase) is required for the activation of Akt-1 by growth factors and insulin. Here we demonstrate by two independent methods that Akt-1 from L6 myotubes binds to PtdIns(3,4,5)P3, PtdIns(3,4)P2 and PtdIns(4,5)P2 when presented against a background of phosphatidylserine (PtdSer) or a 1:1 mixture of PtdSer and phosphatidylcholine (PtdCho). No binding was observed with the lipids PtdIns(3,5)P2, PtdIns4P and PtdIns3P or background lipids. Activated, hyperphosphorylated forms of Akt-1 from insulin-stimulated L6 myotubes bound to PtdIns(3,4,5)P3 in a similar manner as inactive Akt-1. Quantitative analysis using surface plasmon resonance showed that the equilibrium association constant for the binding of Akt-1 to PtdIns(3,4,5)P3 was submicromolar and that PtdIns(3,4)P2 and PtdIns(4,5)P2 bound to Akt-1 with 3- and 6-fold lower affinities respectively. Interaction of Akt-1 with PtdIns(3,4,5)P3 did not activate the protein kinase activity, either before or after incubation with MgATP. A model is presented in which PtdIns(3,4,5)P3 may prime Akt-1 for activation by another protein kinase, perhaps by recruiting it to the plasma membrane. | | 8645147
|
Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction.
Burgering, BM; Coffer, PJ
Nature
376
599-602
1995
显示摘要
A serine/threonine kinase, named protein kinase B (PKB) for its sequence homology to both protein kinase A and C, has previously been isolated. PKB, which is identical to the kinase Rac, was later found to be the cellular homologue of the transforming v-Akt. Here we show that PKB is activated by stimuli such as insulin, platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF). Activation of PKB was inhibited by the phosphatidylinositol-3-OH kinase (PI(3)K) inhibitor wortmannin and by coexpression of a dominant-negative mutant of PI(3)K. PDGF receptor mutants that lack detectable associated PI(3)K activity also fail to induce PKB activation, PKB kinase activity is correlated with phosphorylation of PKB on serine. Finally, we show that a constructed Gag-PKB fusion protein, homologous to the v-akt oncogene, displays significantly increased ligand-independent kinase activity. Furthermore, this activity is sufficient to activate the p70 S6-kinase (p70S6K). These results suggest a role for PKB in PI(3)K-mediated signal transduction. | | 7637810
|