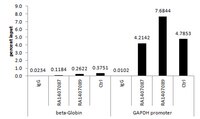
MAF1 represses CDKN1A through a Pol III-dependent mechanism.
Lee, YL; Li, YC; Su, CH; Chiao, CH; Lin, IH; Hsu, MT
eLife
4
e06283
2015
Show Abstract
MAF1 represses Pol III-mediated transcription by interfering with TFIIIB and Pol III. Herein, we found that MAF1 knockdown induced CDKN1A transcription and chromatin looping concurrently with Pol III recruitment. Simultaneous knockdown of MAF1 with Pol III or BRF1 (subunit of TFIIIB) diminished the activation and looping effect, which indicates that recruiting Pol III was required for activation of Pol II-mediated transcription and chromatin looping. Chromatin-immunoprecipitation analysis after MAF1 knockdown indicated enhanced binding of Pol III and BRF1, as well as of CFP1, p300, and PCAF, which are factors that mediate active histone marks, along with the binding of TATA binding protein (TBP) and POLR2E to the CDKN1A promoter. Simultaneous knockdown with Pol III abolished these regulatory events. Similar results were obtained for GDF15. Our results reveal a novel mechanism by which MAF1 and Pol III regulate the activity of a protein-coding gene transcribed by Pol II. | | | 26067234
 |
A comprehensive epigenome map of Plasmodium falciparum reveals unique mechanisms of transcriptional regulation and identifies H3K36me2 as a global mark of gene suppression.
Karmodiya, K; Pradhan, SJ; Joshi, B; Jangid, R; Reddy, PC; Galande, S
Epigenetics & chromatin
8
32
2015
Show Abstract
Role of epigenetic mechanisms towards regulation of the complex life cycle/pathogenesis of Plasmodium falciparum, the causative agent of malaria, has been poorly understood. To elucidate stage-specific epigenetic regulation, we performed genome-wide mapping of multiple histone modifications of P. falciparum. Further to understand the differences in transcription regulation in P. falciparum and its host, human, we compared their histone modification profiles.Our comprehensive comparative analysis suggests distinct mode of transcriptional regulation in malaria parasite by virtue of poised genes and differential histone modifications. Furthermore, analysis of histone modification profiles predicted 562 genes producing anti-sense RNAs and 335 genes having bidirectional promoter activity, which raises the intriguing possibility of RNA-mediated regulation of transcription in P. falciparum. Interestingly, we found that H3K36me2 acts as a global repressive mark and gene regulation is fine tuned by the ratio of activation marks to H3K36me2 in P. falciparum. This novel mechanism of gene regulation is supported by the fact that knockout of SET genes (responsible for H3K36 methylation) leads to up-regulation of genes with highest occupancy of H3K36me2 in wild-type P. falciparum. Moreover, virulence (var) genes are mostly poised and marked by a unique set of activation (H4ac) and repression (H3K9me3) marks, which are mutually exclusive to other Plasmodium housekeeping genes.Our study reveals unique plasticity in the epigenetic regulation in P. falciparum which can influence parasite virulence and pathogenicity. The observed differences in the histone code and transcriptional regulation in P. falciparum and its host will open new avenues for epigenetic drug development against malaria parasite. | | | 26388940
|
Transcriptional regulation of the human TNFSF11 gene in T cells via a cell type-selective set of distal enhancers.
Bishop, KA; Wang, X; Coy, HM; Meyer, MB; Gumperz, JE; Pike, JW
Journal of cellular biochemistry
116
320-30
2015
Show Abstract
In addition to osteoblast lineage cells, the TNF-like factor receptor activator of NF-κB ligand (RANKL) is expressed in both B and T cells and may play a role in bone resorption. Rankl gene (Tnfsf11) expression in mouse T cells is mediated through multiple distal elements marked by increased transcription factor occupancy, histone tail acetylation, and RNA polymerase II recruitment. Little is known, however, of the regulation of human TNFSF11 in T cells. Accordingly, we examined the consequence of T cell activation on the expression of this factor both in Jurkat cells and in primary human T cells. We then explored the mechanism of this regulation by scanning over 400 kb of DNA surrounding the TNFSF11 locus for regulatory enhancers using ChIP-chip analysis. Histone H3/H4 acetylation enrichment identified putative regulatory regions located between -170 and -220 kb upstream of the human TNFSF11 TSS that we designated the human T cell control region (hTCCR). This region showed high sequence conservation with the mouse TCCR. Inhibition of MEK1/2 by U0126 resulted in decreased RANKL expression suggesting that stimulation through MEK1/2 was a prerequisite. ChIP-chip analysis also revealed that c-FOS was recruited to the hTCCR as well. Importantly, both the human TNFSF11 D5a/b (RLD5a/b) enhancer and segments of the hTCCR mediated robust inducible reporter activity following TCR activation. Finally, SNPs implicated in diseases characterized by dysregulated BMD co-localized to the hTCCR region. We conclude that the hTCCR region contains a cell-selective set of enhancers that plays an integral role in the transcriptional regulation of the TNFSF11 gene in human T cells. | | | 25211367
|
Hierarchical clustering of breast cancer methylomes revealed differentially methylated and expressed breast cancer genes.
Lin, IH; Chen, DT; Chang, YF; Lee, YL; Su, CH; Cheng, C; Tsai, YC; Ng, SC; Chen, HT; Lee, MC; Chen, HW; Suen, SH; Chen, YC; Liu, TT; Chang, CH; Hsu, MT
PloS one
10
e0118453
2015
Show Abstract
Oncogenic transformation of normal cells often involves epigenetic alterations, including histone modification and DNA methylation. We conducted whole-genome bisulfite sequencing to determine the DNA methylomes of normal breast, fibroadenoma, invasive ductal carcinomas and MCF7. The emergence, disappearance, expansion and contraction of kilobase-sized hypomethylated regions (HMRs) and the hypomethylation of the megabase-sized partially methylated domains (PMDs) are the major forms of methylation changes observed in breast tumor samples. Hierarchical clustering of HMR revealed tumor-specific hypermethylated clusters and differential methylated enhancers specific to normal or breast cancer cell lines. Joint analysis of gene expression and DNA methylation data of normal breast and breast cancer cells identified differentially methylated and expressed genes associated with breast and/or ovarian cancers in cancer-specific HMR clusters. Furthermore, aberrant patterns of X-chromosome inactivation (XCI) was found in breast cancer cell lines as well as breast tumor samples in the TCGA BRCA (breast invasive carcinoma) dataset. They were characterized with differentially hypermethylated XIST promoter, reduced expression of XIST, and over-expression of hypomethylated X-linked genes. High expressions of these genes were significantly associated with lower survival rates in breast cancer patients. Comprehensive analysis of the normal and breast tumor methylomes suggests selective targeting of DNA methylation changes during breast cancer progression. The weak causal relationship between DNA methylation and gene expression observed in this study is evident of more complex role of DNA methylation in the regulation of gene expression in human epigenetics that deserves further investigation. | | | 25706888
|
Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape.
Veselovska, L; Smallwood, SA; Saadeh, H; Stewart, KR; Krueger, F; Maupetit-Méhouas, S; Arnaud, P; Tomizawa, S; Andrews, S; Kelsey, G
Genome biology
16
209
2015
Show Abstract
Previously, a role was demonstrated for transcription in the acquisition of DNA methylation at imprinted control regions in oocytes. Definition of the oocyte DNA methylome by whole genome approaches revealed that the majority of methylated CpG islands are intragenic and gene bodies are hypermethylated. Yet, the mechanisms by which transcription regulates DNA methylation in oocytes remain unclear. Here, we systematically test the link between transcription and the methylome.We perform deep RNA-Seq and de novo transcriptome assembly at different stages of mouse oogenesis. This reveals thousands of novel non-annotated genes, as well as alternative promoters, for approximately 10 % of reference genes expressed in oocytes. In addition, a large fraction of novel promoters coincide with MaLR and ERVK transposable elements. Integration with our transcriptome assembly reveals that transcription correlates accurately with DNA methylation and accounts for approximately 85-90 % of the methylome. We generate a mouse model in which transcription across the Zac1/Plagl1 locus is abrogated in oocytes, resulting in failure of DNA methylation establishment at all CpGs of this locus. ChIP analysis in oocytes reveals H3K4me2 enrichment at the Zac1 imprinted control region when transcription is ablated, establishing a connection between transcription and chromatin remodeling at CpG islands by histone demethylases.By precisely defining the mouse oocyte transcriptome, this work not only highlights transcription as a cornerstone of DNA methylation establishment in female germ cells, but also provides an important resource for developmental biology research. | | | 26408185
|
Lysine acetyltransferase GCN5b interacts with AP2 factors and is required for Toxoplasma gondii proliferation.
Wang, J; Dixon, SE; Ting, LM; Liu, TK; Jeffers, V; Croken, MM; Calloway, M; Cannella, D; Hakimi, MA; Kim, K; Sullivan, WJ
PLoS pathogens
10
e1003830
2014
Show Abstract
Histone acetylation has been linked to developmental changes in gene expression and is a validated drug target of apicomplexan parasites, but little is known about the roles of individual histone modifying enzymes and how they are recruited to target genes. The protozoan parasite Toxoplasma gondii (phylum Apicomplexa) is unusual among invertebrates in possessing two GCN5-family lysine acetyltransferases (KATs). While GCN5a is required for gene expression in response to alkaline stress, this KAT is dispensable for parasite proliferation in normal culture conditions. In contrast, GCN5b cannot be disrupted, suggesting it is essential for Toxoplasma viability. To further explore the function of GCN5b, we generated clonal parasites expressing an inducible HA-tagged dominant-negative form of GCN5b containing a point mutation that ablates enzymatic activity (E703G). Stabilization of this dominant-negative GCN5b was mediated through ligand-binding to a destabilization domain (dd) fused to the protein. Induced accumulation of the ddHAGCN5b(E703G) protein led to a rapid arrest in parasite replication. Growth arrest was accompanied by a decrease in histone H3 acetylation at specific lysine residues as well as reduced expression of GCN5b target genes in GCN5b(E703G) parasites, which were identified using chromatin immunoprecipitation coupled with microarray hybridization (ChIP-chip). Proteomics studies revealed that GCN5b interacts with AP2-domain proteins, apicomplexan plant-like transcription factors, as well as a "core complex" that includes the co-activator ADA2-A, TFIID subunits, LEO1 polymerase-associated factor (Paf1) subunit, and RRM proteins. The dominant-negative phenotype of ddHAGCN5b(E703G) parasites, considered with the proteomics and ChIP-chip data, indicate that GCN5b plays a central role in transcriptional and chromatin remodeling complexes. We conclude that GCN5b has a non-redundant and indispensable role in regulating gene expression required during the Toxoplasma lytic cycle. | | | 24391497
|
CDK2-dependent phosphorylation of Suv39H1 is involved in control of heterochromatin replication during cell cycle progression.
Park, SH; Yu, SE; Chai, YG; Jang, YK
Nucleic Acids Res
42
6196-207
2014
Show Abstract
Although several studies have suggested that the functions of heterochromatin regulators may be regulated by post-translational modifications during cell cycle progression, regulation of the histone methyltransferase Suv39H1 is not fully understood. Here, we demonstrate a direct link between Suv39H1 phosphorylation and cell cycle progression. We show that CDK2 phosphorylates Suv39H1 at Ser391 and these phosphorylation levels oscillate during the cell cycle, peaking at S phase and maintained during S-G2-M phase. The CDK2-mediated phosphorylation of Suv39H1 at Ser391 results in preferential dissociation from chromatin. Furthermore, phosphorylation-mediated dissociation of Suv39H1 from chromatin causes an enhanced occupancy of JMJD2A histone demethylase on heterochromatin and alterations in inactive histone marks. Overexpression of phospho-mimic Suv39H1 induces early replication of heterochromatin, suggesting the importance of Suv39H1 phosphorylation in the replication of heterochromatin. Moreover, overexpression of phospho-defective Suv39H1 caused altered replication timing of heterochromatin and increases sensitivity to replication stress. Collectively, our data suggest that phosphorylation-mediated modulation of Suv39H1-chromatin association may be an initial step in heterochromatin replication. | | | 24728993
|
A single allele of Hdac2 but not Hdac1 is sufficient for normal mouse brain development in the absence of its paralog.
Hagelkruys, A; Lagger, S; Krahmer, J; Leopoldi, A; Artaker, M; Pusch, O; Zezula, J; Weissmann, S; Xie, Y; Schöfer, C; Schlederer, M; Brosch, G; Matthias, P; Selfridge, J; Lassmann, H; Knoblich, JA; Seiser, C
Development (Cambridge, England)
141
604-16
2014
Show Abstract
The histone deacetylases HDAC1 and HDAC2 are crucial regulators of chromatin structure and gene expression, thereby controlling important developmental processes. In the mouse brain, HDAC1 and HDAC2 exhibit different developmental stage- and lineage-specific expression patterns. To examine the individual contribution of these deacetylases during brain development, we deleted different combinations of Hdac1 and Hdac2 alleles in neural cells. Ablation of Hdac1 or Hdac2 by Nestin-Cre had no obvious consequences on brain development and architecture owing to compensation by the paralog. By contrast, combined deletion of Hdac1 and Hdac2 resulted in impaired chromatin structure, DNA damage, apoptosis and embryonic lethality. To dissect the individual roles of HDAC1 and HDAC2, we expressed single alleles of either Hdac1 or Hdac2 in the absence of the respective paralog in neural cells. The DNA-damage phenotype observed in double knockout brains was prevented by expression of a single allele of either Hdac1 or Hdac2. Strikingly, Hdac1(-/-)Hdac2(+/-) brains showed normal development and no obvious phenotype, whereas Hdac1(+/-)Hdac2(-/-) mice displayed impaired brain development and perinatal lethality. Hdac1(+/-)Hdac2(-/-) neural precursor cells showed reduced proliferation and premature differentiation mediated by overexpression of protein kinase C, delta, which is a direct target of HDAC2. Importantly, chemical inhibition or knockdown of protein kinase C delta was sufficient to rescue the phenotype of neural progenitor cells in vitro. Our data indicate that HDAC1 and HDAC2 have a common function in maintaining proper chromatin structures and show that HDAC2 has a unique role by controlling the fate of neural progenitors during normal brain development. | Immunohistochemistry | | 24449838
|
Dynamic remodeling of histone modifications in response to osmotic stress in Saccharomyces cerevisiae.
Magraner-Pardo, L; Pelechano, V; Coloma, MD; Tordera, V
BMC genomics
15
247
2014
Show Abstract
Specific histone modifications play important roles in chromatin functions; i.e., activation or repression of gene transcription. This participation must occur as a dynamic process. Nevertheless, most of the histone modification maps reported to date provide only static pictures that link certain modifications with active or silenced states. This study, however, focuses on the global histone modification variation that occurs in response to the transcriptional reprogramming produced by a physiological perturbation in yeast.We did a genome-wide chromatin immunoprecipitation analysis for eight specific histone modifications before and after saline stress. The most striking change was rapid acetylation loss in lysines 9 and 14 of H3 and in lysine 8 of H4, associated with gene repression. The genes activated by saline stress increased the acetylation levels at these same sites, but this acetylation process was quantitatively minor if compared to that of the deacetylation of repressed genes. The changes in the tri-methylation of lysines 4, 36 and 79 of H3 and the di-methylation of lysine 79 of H3 were slighter than those of acetylation. Furthermore, we produced new genome-wide maps for seven histone modifications, and we analyzed, for the first time in S. cerevisiae, the genome-wide profile of acetylation of lysine 8 of H4.This research reveals that the short-term changes observed in the post-stress methylation of histones are much more moderate than those of acetylation, and that the dynamics of the acetylation state of histones during activation or repression of transcription is a much quicker process than methylation. | | | 24678875
|
Radiation-induced alterations of histone post-translational modification levels in lymphoblastoid cell lines.
Maroschik, B; Gürtler, A; Krämer, A; Rößler, U; Gomolka, M; Hornhardt, S; Mörtl, S; Friedl, AA
Radiation oncology (London, England)
9
15
2014
Show Abstract
Radiation-induced alterations in posttranslational histone modifications (PTMs) may affect the cellular response to radiation damage in the DNA. If not reverted appropriately, altered PTM patterns may cause long-term alterations in gene expression regulation and thus lead to cancer. It is therefore important to characterize radiation-induced alterations in PTM patterns and the factors affecting them.A lymphoblastoid cell line established from a normal donor was used to screen for alterations in methylation levels at H3K4, H3K9, H3K27, and H4K20, as well as acetylation at H3K9, H3K56, H4K5, and H4K16, by quantitative Western Blot analysis at 15 min, 1 h and 24 h after irradiation with 2 Gy and 10 Gy. The variability of alterations in acetylation marks was in addition investigated in a panel of lymphoblastoid cell lines with differing radiosensitivity established from lung cancer patients.The screening procedure demonstrated consistent hypomethylation at H3K4me3 and hypoacetylation at all acetylation marks tested. In the panel of lymphoblastoid cell lines, however, a high degree of inter-individual variability became apparent. Radiosensitive cell lines showed more pronounced and longer lasting H4K16 hypoacetylation than radioresistant lines, which correlates with higher levels of residual γ-H2AX foci after 24 h.So far, the factors affecting extent and duration of radiation-induced histone alterations are poorly defined. The present work hints at a high degree of inter-individual variability and a potential correlation of DNA damage repair capacity and alterations in PTM levels. | Western Blotting | | 24406105
|