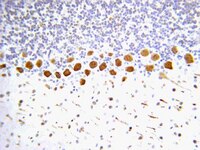
Pleiotropic Effects of Levofloxacin, Fluoroquinolone Antibiotics, against Influenza Virus-Induced Lung Injury.
Enoki, Y; Ishima, Y; Tanaka, R; Sato, K; Kimachi, K; Shirai, T; Watanabe, H; Chuang, VT; Fujiwara, Y; Takeya, M; Otagiri, M; Maruyama, T
PloS one
10
e0130248
2015
Afficher le résumé
Reactive oxygen species (ROS) and nitric oxide (NO) are major pathogenic molecules produced during viral lung infections, including influenza. While fluoroquinolones are widely used as antimicrobial agents for treating a variety of bacterial infections, including secondary infections associated with the influenza virus, it has been reported that they also function as anti-oxidants against ROS and as a NO regulator. Therefore, we hypothesized that levofloxacin (LVFX), one of the most frequently used fluoroquinolone derivatives, may attenuate pulmonary injuries associated with influenza virus infections by inhibiting the production of ROS species such as hydroxyl radicals and neutrophil-derived NO that is produced during an influenza viral infection. The therapeutic impact of LVFX was examined in a PR8 (H1N1) influenza virus-induced lung injury mouse model. ESR spin-trapping experiments indicated that LVFX showed scavenging activity against neutrophil-derived hydroxyl radicals. LVFX markedly improved the survival rate of mice that were infected with the influenza virus in a dose-dependent manner. In addition, the LVFX treatment resulted in a dose-dependent decrease in the level of 8-hydroxy-2'-deoxyguanosine (a marker of oxidative stress) and nitrotyrosine (a nitrative marker) in the lungs of virus-infected mice, and the nitrite/nitrate ratio (NO metabolites) and IFN-γ in BALF. These results indicate that LVFX may be of substantial benefit in the treatment of various acute inflammatory disorders such as influenza virus-induced pneumonia, by inhibiting inflammatory cell responses and suppressing the overproduction of NO in the lungs. | | 26086073
 |
Exposure to 100% Oxygen Abolishes the Impairment of Fracture Healing after Thoracic Trauma.
Kemmler, J; Bindl, R; McCook, O; Wagner, F; Gröger, M; Wagner, K; Scheuerle, A; Radermacher, P; Ignatius, A
PloS one
10
e0131194
2015
Afficher le résumé
In polytrauma patients a thoracic trauma is one of the most critical injuries and an important trigger of post-traumatic inflammation. About 50% of patients with thoracic trauma are additionally affected by bone fractures. The risk for fracture malunion is considerably increased in such patients, the pathomechanisms being poorly understood. Thoracic trauma causes regional alveolar hypoxia and, subsequently, hypoxemia, which in turn triggers local and systemic inflammation. Therefore, we aimed to unravel the role of oxygen in impaired bone regeneration after thoracic trauma. We hypothesized that short-term breathing of 100% oxygen in the early post-traumatic phase ameliorates inflammation and improves bone regeneration. Mice underwent a femur osteotomy alone or combined with blunt chest trauma 100% oxygen was administered immediately after trauma for two separate 3 hour intervals. Arterial blood gas tensions, microcirculatory perfusion and oxygenation were assessed at 3, 9 and 24 hours after injury. Inflammatory cytokines and markers of oxidative/nitrosative stress were measured in plasma, lung and fracture hematoma. Bone healing was assessed on day 7, 14 and 21. Thoracic trauma induced pulmonary and systemic inflammation and impaired bone healing. Short-term exposure to 100% oxygen in the acute post-traumatic phase significantly attenuated systemic and local inflammatory responses and improved fracture healing without provoking toxic side effects, suggesting that hyperoxia could induce anti-inflammatory and pro-regenerative effects after severe injury. These results suggest that breathing of 100% oxygen in the acute post-traumatic phase might reduce the risk of poorly healing fractures in severely injured patients. | | 26147725
|
Ferrous sulfate, but not iron polymaltose complex, aggravates local and systemic inflammation and oxidative stress in dextran sodium sulfate-induced colitis in rats.
Toblli, JE; Cao, G; Angerosa, M
Drug design, development and therapy
9
2585-97
2015
Afficher le résumé
Iron deficiency is common in inflammatory bowel disease, yet oral iron therapy may worsen the disease symptoms and increase systemic and local oxidative stress. The aim of this study was to compare the effects of oral ferrous sulfate and iron polymaltose complex on inflammatory and oxidative stress markers in colitic rats.Animals were divided into four groups with ten animals each. Rats of three groups received dextran sodium sulfate to induce colitis and animals of two of these groups received 5 mg iron/kg of body weight a day, as ferrous sulfate or iron polymaltose complex, for 7 days. Gross colon anatomy, histology of colon and liver, stainings of L-ferritin, Prussian blue, hepcidin, tumor necrosis factor-α, and interleukin-6, as well serum levels of liver enzymes, inflammatory markers, and iron markers, were assessed.Body weight, gross anatomy, crypt injury and inflammation scores, inflammatory parameters in liver and colon, as well as serum and liver hepcidin levels were not significantly different between colitic animals without iron treatment and colitic animals treated with iron polymaltose complex. In contrast, ferrous sulfate treatment caused significant worsening of these parameters. As opposed to ferrous sulfate, iron polymaltose complex caused less or no additional oxidative stress in the colon and liver compared to colitic animals without iron treatment.Iron polymaltose complex had negligible effects on colonic tissue erosion, local or systemic oxidative stress, and local or systemic inflammation, even at high therapeutic doses, and may thus represent a valuable oral treatment of iron deficiency in inflammatory bowel disease. | | 26005335
|
Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress.
Ikeda, M; Ide, T; Fujino, T; Arai, S; Saku, K; Kakino, T; Tyynismaa, H; Yamasaki, T; Yamada, K; Kang, D; Suomalainen, A; Sunagawa, K
PloS one
10
e0119687
2015
Afficher le résumé
Mitochondrial DNA (mtDNA) copy number decreases in animal and human heart failure (HF), yet its role in cardiomyocytes remains to be elucidated. Thus, we investigated the cardioprotective function of increased mtDNA copy number resulting from the overexpression of human transcription factor A of mitochondria (TFAM) or Twinkle helicase in volume overload (VO)-induced HF.Two strains of transgenic (TG) mice, one overexpressing TFAM and the other overexpressing Twinkle helicase, exhibit an approximately 2-fold equivalent increase in mtDNA copy number in heart. These TG mice display similar attenuations in eccentric hypertrophy and improved cardiac function compared to wild-type (WT) mice without any deterioration of mitochondrial enzymatic activities in response to VO, which was accompanied by a reduction in matrix-metalloproteinase (MMP) activity and reactive oxygen species after 8 weeks of VO. Moreover, acute VO-induced MMP-2 and MMP-9 upregulation was also suppressed at 24 h in both TG mice. In isolated rat cardiomyocytes, mitochondrial reactive oxygen species (mitoROS) upregulated MMP-2 and MMP-9 expression, and human TFAM (hTFAM) overexpression suppressed mitoROS and their upregulation. Additionally, mitoROS were equally suppressed in H9c2 rat cardiomyoblasts that overexpress hTFAM or rat Twinkle, both of which exhibit increased mtDNA copy number. Furthermore, mitoROS and mitochondrial protein oxidation from both TG mice were suppressed compared to WT mice.The overexpression of TFAM or Twinkle results in increased mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. Our findings suggest that increasing mtDNA copy number could be a useful therapeutic strategy to target mitoROS in HF. | | 25822152
|
Ferric carboxymaltose-mediated attenuation of Doxorubicin-induced cardiotoxicity in an iron deficiency rat model.
Toblli, JE; Rivas, C; Cao, G; Giani, JF; Funk, F; Mizzen, L; Dominici, FP
Chemotherapy research and practice
2014
570241
2014
Afficher le résumé
Since anthracycline-induced cardiotoxicity (AIC), a complication of anthracycline-based chemotherapies, is thought to involve iron, concerns exist about using iron for anaemia treatment in anthracycline-receiving cancer patients. This study evaluated how intravenous ferric carboxymaltose (FCM) modulates the influence of iron deficiency anaemia (IDA) and doxorubicin (3-5 mg per kg body weight [BW]) on oxidative/nitrosative stress, inflammation, and cardiorenal function in spontaneously hypertensive stroke-prone (SHR-SP) rats. FCM was given as repeated small or single total dose (15 mg iron per kg BW), either concurrent with or three days after doxorubicin. IDA (after dietary iron restriction) induced cardiac and renal oxidative stress (markers included malondialdehyde, catalase, Cu,Zn-superoxide dismutase, and glutathione peroxidase), nitrosative stress (inducible nitric oxide synthase and nitrotyrosine), inflammation (tumour necrosis factor-alpha and interleukin-6), and functional/morphological abnormalities (left ventricle end-diastolic and end-systolic diameter, fractional shortening, density of cardiomyocytes and capillaries, caveolin-1 expression, creatinine clearance, and urine neutrophil gelatinase-associated lipocalin) that were aggravated by doxorubicin. Notably, iron treatment with FCM did not exacerbate but attenuated the cardiorenal effects of IDA and doxorubicin independent of the iron dosing regimen. The results of this model suggest that intravenous FCM can be used concomitantly with an anthracycline-based chemotherapy without increasing signs of AIC. | | 24876963
|
Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity.
Rivera, JF; Costes, S; Gurlo, T; Glabe, CG; Butler, PC
The Journal of clinical investigation
124
3489-500
2014
Afficher le résumé
Type 2 diabetes (T2D) is characterized by a deficiency in β cell mass, increased β cell apoptosis, and extracellular accumulation of islet amyloid derived from islet amyloid polypeptide (IAPP), which β cells coexpress with insulin. IAPP expression is increased in the context of insulin resistance, the major risk factor for developing T2D. Human IAPP is potentially toxic, especially as membrane-permeant oligomers, which have been observed to accumulate within β cells of patients with T2D and rodents expressing human IAPP. Here, we determined that β cell IAPP content is regulated by autophagy through p62-dependent lysosomal degradation. Induction of high levels of human IAPP in mouse β cells resulted in accumulation of this amyloidogenic protein as relatively inert fibrils within cytosolic p62-positive inclusions, which temporarily averts formation of toxic oligomers. Mice hemizygous for transgenic expression of human IAPP did not develop diabetes; however, loss of β cell-specific autophagy in these animals induced diabetes, which was attributable to accumulation of toxic human IAPP oligomers and loss of β cell mass. In human IAPP-expressing mice that lack β cell autophagy, increased oxidative damage and loss of an antioxidant-protective pathway appeared to contribute to increased β cell apoptosis. These findings indicate that autophagy/lysosomal degradation defends β cells against proteotoxicity induced by oligomerization-prone human IAPP. | | 25036708
|
Therapeutic impact of human serum albumin-thioredoxin fusion protein on influenza virus-induced lung injury mice.
Tanaka, R; Ishima, Y; Enoki, Y; Kimachi, K; Shirai, T; Watanabe, H; Chuang, VT; Maruyama, T; Otagiri, M
Frontiers in immunology
5
561
2014
Afficher le résumé
Reactive oxygen species (ROS) are the primary pathogenic molecules produced in viral lung infections. We previously reported on the use of a recombinant human serum albumin (HSA)-thioredoxin 1 (Trx) fusion protein (HSA-Trx) for extending the half-life Trx, an endogenous protein with anti-oxidant properties. As a result, it was possible to overcome the unfavorable pharmacokinetic and short pharmacological properties of Trx. We hypothesized that HSA-Trx would attenuate the enhanced ROS production of species such as hydroxyl radicals by neutrophils during an influenza viral infection. The levels of 8-hydroxy-2'-deoxyguanosine and 3-nitrotyrosine were used as indices of the anti-oxidant activity of HSA-Trx. In addition, the cytoprotective effects of HSA-Trx were examined in PR8 (H1N1) influenza virus-induced lung injured mice. The findings show that HSA-Trx reduced the number of total cells, neutrophils, and total protein in BALF of influenza virus-induced lung injured mice. The HSA-Trx treatment significantly decreased the level of 8-hydroxy-2'-deoxyguanosine and 3-nitrotyrosine, but failed to inhibit inducible nitric oxide synthase expression, in the lungs of the virus-infected mice. On the other hand, Tamiflu(®) treatment also significantly suppressed the production of inflammatory cells and neutrophil infiltration, as well as the protein level in BALF and lung histopathological alterations caused by the influenza virus. The suppressive effect of Tamiflu(®) was slightly stronger than that of HSA-Trx. Interestingly, Tamiflu(®) significantly decreased virus proliferation, while HSA-Trx had no effect. These results indicate that HSA-Trx may be of therapeutic value for the treatment of various acute inflammatory disorders such as influenza-virus-induced pneumonia, by inhibiting inflammatory-cell responses and suppressing the overproduction of NO in the lung. | | 25414704
|
Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease.
Galloway, CA; Lee, H; Brookes, PS; Yoon, Y
American journal of physiology. Gastrointestinal and liver physiology
307
G632-41
2014
Afficher le résumé
Mitochondria produce the majority of cellular ATP through oxidative phosphorylation, and their capacity to do so is influenced by many factors. Mitochondrial morphology is recently suggested as an important contributor in controlling mitochondrial bioenergetics. Mitochondria divide and fuse continuously, which is affected by environmental factors, including metabolic alterations. Underscoring its bioenergetic influence, altered mitochondrial morphology is reported in tissues of patients and in animal models of metabolic dysfunction. In this study, we found that mitochondrial fission plays a vital role in the progression of nonalcoholic fatty liver disease (NAFLD). The development of hepatic steatosis, oxidative/nitrative stress, and hepatic tissue damage, induced by a high-fat diet, were alleviated in genetically manipulated mice suppressing mitochondrial fission. The alleviation of steatosis was recapitulated in primary hepatocytes with the inhibition of mitochondrial fission. Mechanistically, our study indicates that fission inhibition enhances proton leak under conditions of free fatty acid incubation, implicating bioenergetic change through manipulating mitochondrial fission. Taken together, our results suggest a mechanistic role for mitochondrial fission in the etiology of NAFLD. The efficacy of decreasing mitochondrial fission in the suppression of NAFLD suggests that mitochondrial fission represents a novel target for therapeutic treatment of NAFLD. | | 25080922
|
Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male Zucker obese rats.
Aroor, AR; Sowers, JR; Bender, SB; Nistala, R; Garro, M; Mugerfeld, I; Hayden, MR; Johnson, MS; Salam, M; Whaley-Connell, A; Demarco, VG
Endocrinology
154
2501-13
2013
Afficher le résumé
Diastolic dysfunction is a prognosticator for future cardiovascular events that demonstrates a strong correlation with obesity. Pharmacological inhibition of dipeptidylpeptidase-4 (DPP-4) to increase the bioavailability of glucagon-like peptide-1 is an emerging therapy for control of glycemia in type 2 diabetes patients. Accumulating evidence suggests that glucagon-like peptide-1 has insulin-independent actions in cardiovascular tissue. However, it is not known whether DPP-4 inhibition improves obesity-related diastolic dysfunction. Eight-week-old Zucker obese (ZO) and Zucker lean rats were fed normal chow diet or diet containing the DPP-4 inhibitor, linagliptin (LGT), for 8 weeks. Plasma DPP-4 activity was 3.3-fold higher in ZO compared with Zucker lean rats and was reduced by 95% with LGT treatment. LGT improved echocardiographic and pressure volume-derived indices of diastolic function that were impaired in ZO control rats, without altering food intake or body weight gain during the study period. LGT also blunted elevated blood pressure progression in ZO rats involving improved skeletal muscle arteriolar function, without reducing left ventricular hypertrophy, fibrosis, or oxidative stress in ZO hearts. Expression of phosphorylated- endothelial nitric oxide synthase (eNOS)(Ser1177), total eNOS, and sarcoplasmic reticulum calcium ATPase 2a protein was elevated in the LGT-treated ZO heart, suggesting improved Ca(2+) handling. The ZO myocardium had an abnormal mitochondrial sarcomeric arrangement and cristae structure that were normalized by LGT. These studies suggest that LGT reduces blood pressure and improves intracellular Cai(2+) mishandling and cardiomyocyte ultrastructure, which collectively result in improvements in diastolic function in the absence of reductions in left ventricular hypertrophy, fibrosis, or oxidative stress in insulin-resistant ZO rats. | | 23653460
|
Status epilepticus induces vasogenic edema via tumor necrosis factor-α/ endothelin-1-mediated two different pathways.
Kim, JE; Ryu, HJ; Kang, TC
PloS one
8
e74458
2013
Afficher le résumé
Status epilepticus (SE) induces vasogenic edema in the piriform cortex with disruptions of the blood-brain barrier (BBB). However, the mechanisms of vasogenic edema formation following SE are still unknown. Here we investigated the endothelin B (ETB) receptor-mediated pathway of SE-induced vasogenic edema. Following SE, the release of tumor necrosis factor-α (TNF-α) stimulated endothelin-1 (ET-1) release and expression in neurons and endothelial cells. In addition, TNF-α-induced ET-1 increased BBB permeability via ETB receptor-mediated endothelial nitric oxide synthase (eNOS) activation in endothelial cells. ETB receptor activation also increased intracellular reactive oxygen species by NADPH oxidase production in astrocytes. These findings suggest that SE results in BBB dysfunctions via endothelial-astroglial interactions through the TNF-α-ET-1-eNOS/NADPH oxidase pathway, and that these ETB receptor-mediated interactions may be an effective therapeutic strategy for vasogenic edema in various neurological diseases. | | 24040253
|