Inhibition of cyclin-dependent kinase 5 but not of glycogen synthase kinase 3-β prevents neurite retraction and tau hyperphosphorylation caused by secretable products of human T-cell leukemia virus type I-infected lymphocytes.
Maldonado, H; Ramírez, E; Utreras, E; Pando, ME; Kettlun, AM; Chiong, M; Kulkarni, AB; Collados, L; Puente, J; Cartier, L; Valenzuela, MA
Journal of neuroscience research
89
1489-98
2010
Mostrar resumen
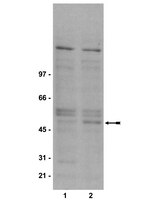
Human T-cell leukemia virus type I (HTLV-I)-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is a neurodegenerative disease characterized by selective loss of axons and myelin in the corticospinal tracts. This central axonopathy may originate from the impairment of anterograde axoplasmic transport. Previous work showed tau hyperphosphorylation at T(181) in cerebrospinal fluid of HAM/TSP patients. Similar hyperphosphorylation occurs in SH-SY5Y cells incubated with supernatant from MT-2 cells (HTLV-I-infected lymphocytes secreting viral proteins, including Tax) that produce neurite shortening. Tau phosphorylation at T(181) is attributable to glycogen synthase kinase 3-β (GSK3-β) and cyclin-dependent kinase 5 (CDK5) activation. Here we investigate whether neurite retraction in the SH-SY5Y model associates with concurrent changes in other tau hyperphosphorylable residues. Threonine 181 turned out to be the only tau hyperphosphorylated residue. We also evaluate the role of GSK3-β and CDK5 in this process by using specific kinase inhibitors (LiCl, TDZD-8, and roscovitine). Changes in both GSK3-β active and inactive forms were followed by measuring the regulatory phosphorylable sites (S(9) and Y(216) , inactivating and activating phosphorylation, respectively) together with changes in β-catenin protein levels. Our results showed that LiCl and TDZD-8 were unable to prevent MT-2 supernatant-mediated neurite retraction and also that neither Y(216) nor S(9) phosphorylations were changed in GSK3-β. Thus, GSK3-β seems not to play a role in T(181) hyperphosphorylation. On the other hand, the CDK5 involvement in tau phosphorylation was confirmed by both the increase in its enzymatic activity and the absence of MT-2 neurite retraction in the presence of roscovitine or CDK5 siRNA transfection. | | 21671254
 |
Measuring GSK3 expression and activity in cells.
Adam R Cole,Calum Sutherland
Methods in molecular biology (Clifton, N.J.)
468
2008
Mostrar resumen
Glycogen synthase kinase (GSK)-3 is a key signalling intermediate in the action ofWnts. This protein kinase is ubiquitously expressed and has high inherent activity but is inhibited by activation of Wnt signalling or activation of growth factor receptor tyrosine kinases (e.g. insulin, nerve growth factor [NGF], platelet-derived growth factor [PDGF], etc.). The degree of inhibition of GSK3 in cells treated with such reagents is dependent on the cell type and the stimulus used. Therefore, the ability to accurately measure GSK3 activity in cells is an important aspect of GSK3 research. The activity of GSK3 is reduced by posttranslational modification (phosphorylation) and this can be measured by immunoblot with specific reagents (indirect), or by immunoprecipitation and assay (direct), as long as the modification is protected during these procedures. However, inhibition by phosphorylation is specific to cellular activation by growth factors and nutrients. Wnt inhibition of GSK3 does not involve phosphorylation of these residues on GSK3 and therefore it cannot be measured using this modification. Currently, the simplest way to assess Wnt inhibition of GSK3 is to monitor phosphorylation of specific GSK3 substrates in cells (e.g. beta-catenin). Alternatively, Wnt inhibition of GSK3 can be measured by partial purification of cellular GSK3 by ion exchange chromatography and assay of fractions or possibly by immunoprecipitation and assay. In this chapter, we demonstrate the use of the different approaches to measure GSK3 activity in SH-SY5Y cells, describe the best antibodies currently available, and discuss the potential drawbacks of each method. | | 19099245
|
Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation.
Pruitt, K; Zinn, RL; Ohm, JE; McGarvey, KM; Kang, SH; Watkins, DN; Herman, JG; Baylin, SB
PLoS genetics
2
e40
2005
Mostrar resumen
The class III histone deactylase (HDAC), SIRT1, has cancer relevance because it regulates lifespan in multiple organisms, down-regulates p53 function through deacetylation, and is linked to polycomb gene silencing in Drosophila. However, it has not been reported to mediate heterochromatin formation or heritable silencing for endogenous mammalian genes. Herein, we show that SIRT1 localizes to promoters of several aberrantly silenced tumor suppressor genes (TSGs) in which 5' CpG islands are densely hypermethylated, but not to these same promoters in cell lines in which the promoters are not hypermethylated and the genes are expressed. Heretofore, only type I and II HDACs, through deactylation of lysines 9 and 14 of histone H3 (H3-K9 and H3-K14, respectively), had been tied to the above TSG silencing. However, inhibition of these enzymes alone fails to re-activate the genes unless DNA methylation is first inhibited. In contrast, inhibition of SIRT1 by pharmacologic, dominant negative, and siRNA (small interfering RNA)-mediated inhibition in breast and colon cancer cells causes increased H4-K16 and H3-K9 acetylation at endogenous promoters and gene re-expression despite full retention of promoter DNA hypermethylation. Furthermore, SIRT1 inhibition affects key phenotypic aspects of cancer cells. We thus have identified a new component of epigenetic TSG silencing that may potentially link some epigenetic changes associated with aging with those found in cancer, and provide new directions for therapeutically targeting these important genes for re-expression. | Western Blotting | 16596166
|