Lipolysaccharide-Induced Neuroinflammation Is Associated with Alzheimer-Like Amyloidogenic Axonal Pathology and Dendritic Degeneration in Rats.
Deng, X; Li, M; Ai, W; He, L; Lu, D; Patrylo, PR; Cai, H; Luo, X; Li, Z; Yan, X
Advances in Alzheimer's disease
3
78-93
2014
Mostrar resumen
Chronic neuroinflammation is thought to play an etiological role in Alzheimer's disease (AD), which is characterized pathologically by amyloid and tau formation, as well as neuritic dystrophy and synaptic degeneration. The causal relationship between these pathological events is a topic of ongoing research and discussion. Recent data from transgenic AD models point to a tight spatiotemporal link between neuritic and amyloid pathology, with the obligatory enzyme for β-amyloid (Aβ) production, namely β-secretase-1 (BACE1), is overexpressed in axon terminals undergoing dystrophic change. However, the axonal pathology inherent with BACE1 elevation seen in transgenic AD mice may be secondary to increased soluble Aβ in these genetically modified animals. Here we explored the occurrence of the AD-like axonal and dendritic pathology in adult rat brain affected by LPS-induced chronic neuroinflammation. Unilateral intracerebral LPS injection induced prominent inflammatory response in glial cells in the ipsilateral cortex and hippocampal formation. BACE1 protein levels were elevated the ipsilateral hippocampal lysates in the LPS treated animals relative to controls. BACE1 immunoreactive dystrophic axons appeared in the LPS-treated ipsilateral cortex and hippocampal formation, colocalizing with increased β-amyloid precursor protein and Aβ antibody (4G8) immunolabeling. Quantitative Golgi studies revealed reduction of dendritic branching points and spine density on cortical layer III and hippocampal CA3 pyramidal neurons in the LPS-treated ipsilateral cerebrum. These findings suggest that Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration occur in wildtype mammalian brain in partnership with neuroinflammation following LPS injection. | | | 25360394
 |
Amyloid plaque pathogenesis in 5XFAD mouse spinal cord: retrograde transneuronal modulation after peripheral nerve injury.
Li, JM; Xue, ZQ; Deng, SH; Luo, XG; Patrylo, PR; Rose, GW; Cai, H; Cai, Y; Yan, XX
Neurotoxicity research
24
1-14
2013
Mostrar resumen
The spinal cord is composed of distinct neuronal groups with well-defined anatomic connections. In some transgenic (Tg) models of Alzheimer's disease (AD), amyloid plaques develop in this structure, although the underlying cellular mechanism remains elusive. We attempted to explore the origin, evolution, and modulation of spinal β-amyloid (Aβ) deposition using Tg mice harboring five familiar AD-related mutations (5XFAD) as an experiential model. Dystrophic neuritic elements with enhanced β-secretase-1 (BACE1) immunoreactivity (IR) appeared as early as 2 months of age, and increased with age up to 12 months examined in this study, mostly over the ventral horn (VH). Extracellular Aβ IR emerged and developed during this same period, site-specifically co-existing with BACE1-labeled neurites often in the vicinity of large VH neurons that expressed the mutant human APP. The BACE1-labeled neurites almost invariably colocalized with β-amyloid precursor protein (APP) and synaptophysin, and frequently with the vesicular glutamate transporter-1 (VGLUT). Reduced IR for the neuronal-specific nuclear antigen (NeuN) occurred in the VH by 12 months of age. In 8-month-old animals surviving 6 months after a unilateral sciatic nerve transection, there were significant increases of Aβ, BACE1, and VGLUT IR in the VN of the ipsilateral relative to contralateral lumbar spinal segments. These results suggest that extracellular Aβ deposition in 5XFAD mouse spinal cord relates to a progressive and amyloidogenic synaptic pathology largely involving presynaptic axon terminals from projection neurons in the brain. Spinal neuritic plaque formation is enhanced after peripheral axotomy, suggesting a retrograde transneuronal modulation on pathogenesis. | | | 23055086
|
Lidocaine attenuates cognitive impairment after isoflurane anesthesia in old rats.
Lin, D; Cao, L; Wang, Z; Li, J; Washington, JM; Zuo, Z
Behavioural brain research
228
319-27
2011
Mostrar resumen
Post-operative cognitive dysfunction (POCD) is a clinical phenomenon that has drawn significant attention from the public and scientific community. Age is a risk factor for POCD. However, the contribution of general anesthesia/anesthetics to POCD and the underlying neuropathology are not clear. Here, we showed that 18-month-old male Fisher 344 rats exposed to 1.2% isoflurane, a general anesthetic, for 2h had significant learning and memory impairments assessed at 2-4 weeks after isoflurane exposure. These isoflurane effects were attenuated by intravenous lidocaine (1.5mg/kg as a bolus and then 2mg/kg/h during isoflurane exposure), a local anesthetic that has neuroprotective effect. Exposure to isoflurane or isoflurane plus lidocaine did not change the neuronal and synaptic density as well as the expression of NeuN (a neuronal protein), drebrin (a dendritic spine protein), synaptophysin (a synaptic protein), activated caspase 3 and caspase-activated DNase in the hippocampus at 29 days after isoflurane exposure when cognitive impairment was present. Isoflurane and lidocaine did not affect the amount of β-amyloid peptide, total tau and phospho-tau in the cerebral cortex as well as interleukin-1β and tumor necrosis factor-α in the hippocampus at 29 days after isoflurane exposure. Thus, isoflurane induces learning and memory impairment in old rats. Lidocaine attenuates these isoflurane effects. Isoflurane may not cause long-lasting neuropathological changes. | | | 22192381
|
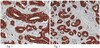
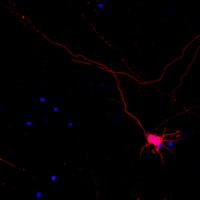
BACE1 elevation is involved in amyloid plaque development in the triple transgenic model of Alzheimer's disease: differential Aβ antibody labeling of early-onset axon terminal pathology.
Cai, Y; Zhang, XM; Macklin, LN; Cai, H; Luo, XG; Oddo, S; Laferla, FM; Struble, RG; Rose, GM; Patrylo, PR; Yan, XX
Neurotoxicity research
21
160-74
2011
Mostrar resumen
β-amyloid precursor protein (APP) and presenilins mutations cause early-onset familial Alzheimer's disease (FAD). Some FAD-based mouse models produce amyloid plaques, others do not. β-Amyloid (Aβ) deposition can manifest as compact and diffuse plaques; it is unclear why the same Aβ molecules aggregate in different patterns. Is there a basic cellular process governing Aβ plaque pathogenesis? We showed in some FAD mouse models that compact plaque formation is associated with a progressive axonal pathology inherent with increased expression of β-secretase (BACE1), the enzyme initiating the amyloidogenic processing of APP. A monoclonal Aβ antibody, 3D6, visualized distinct axon terminal labeling before plaque onset. The present study was set to understand BACE1 and axonal changes relative to diffuse plaque development and to further characterize the novel axonal Aβ antibody immunoreactivity (IR), using triple transgenic AD (3xTg-AD) mice as experimental model. Diffuse-like plaques existed in the forebrain in aged transgenics and were regionally associated with increased BACE1 labeled swollen/sprouting axon terminals. Increased BACE1/3D6 IR at axon terminals occurred in young animals before plaque onset. These axonal elements were also co-labeled by other antibodies targeting the N-terminal and mid-region of Aβ domain and the C-terminal of APP, but not co-labeled by antibodies against the Aβ C-terminal and APP N-terminal. The results suggest that amyloidogenic axonal pathology precedes diffuse plaque formation in the 3xTg-AD mice, and that the early-onset axonal Aβ antibody IR in transgenic models of AD might relate to a cross-reactivity of putative APP β-carboxyl terminal fragments. | Immunohistochemistry | Mouse | 21725719
|
Functional deprivation promotes amyloid plaque pathogenesis in Tg2576 mouse olfactory bulb and piriform cortex.
Zhang, XM; Xiong, K; Cai, Y; Cai, H; Luo, XG; Feng, JC; Clough, RW; Patrylo, PR; Struble, RG; Yan, XX
The European journal of neuroscience
31
710-21
2009
Mostrar resumen
Cerebral hypometabolism and amyloid accumulation are principal neuropathological manifestations of Alzheimer's disease (AD). Whether and how brain/neuronal activity might modulate certain pathological processes of AD are interesting topics of recent clinical and basic research in the field, and may be of potential medical relevance in regard to both the disease etiology and intervention. Using the Tg2576 transgenic mouse model of AD, this study characterized a promotive effect of neuronal hypoactivity associated with functional deprivation on amyloid plaque pathogenesis in the olfactory pathway. Unilateral naris-occlusion caused beta-secretase-1 (BACE1) elevation in neuronal terminals in the deprived relative to the non-deprived bulb and piriform cortex in young adult mice. In parallel with the overall age-related plaque development in the forebrain, locally increased BACE1 immunoreactivity co-occurred with amyloid deposition first in the piriform cortex then within the bulb, more prominent on the deprived relative to the non-deprived side. Biochemical analyses confirmed elevated BACE1 protein levels, enzymatic activity and products in the deprived relative to non-deprived bulbs. Plaque-associated BACE1 immunoreactivity in the bulb and piriform cortex was localized preferentially to swollen/sprouting glutamatergic axonal terminals, with Abeta immunoreactivity occurring inside as well as around these terminals. Together, these findings suggest that functional deprivation or neuronal hypoactivity facilitates amyloid plaque formation in the forebrain in a transgenic model of AD, which operates synergistically with age effect. The data also implicate an intrinsic association of amyloid accumulation and plaque formation with progressive axonal pathology. Artículo Texto completo | Immunohistochemistry | Mouse | 20384814
|
β-Secretase-1 elevation in aged monkey and Alzheimer's disease human cerebral cortex occurs around the vasculature in partnership with multisystem axon terminal pathogenesis and β-amyloid accumulation.
Cai, Y; Xiong, K; Zhang, XM; Cai, H; Luo, XG; Feng, JC; Clough, RW; Struble, RG; Patrylo, PR; Chu, Y; Kordower, JH; Yan, XX
The European journal of neuroscience
32
1223-38
2009
Mostrar resumen
Alzheimer's disease (AD) is the most common dementia-causing disorder in the elderly; it may be related to multiple risk factors, and is characterized pathologically by cerebral hypometabolism, paravascular β-amyloid peptide (Aβ) plaques, neuritic dystrophy, and intra-neuronal aggregation of phosphorylated tau. To explore potential pathogenic links among some of these lesions, we examined β-secretase-1 (BACE1) alterations relative to Aβ deposition, neuritic pathology and vascular organization in aged monkey and AD human cerebral cortex. Western blot analyses detected increased levels of BACE1 protein and β-site-cleavage amyloid precursor protein C-terminal fragments in plaque-bearing human and monkey cortex relative to controls. In immunohistochemistry, locally elevated BACE1 immunoreactivity (IR) occurred in AD but not in control human cortex, with a trend for increased overall density among cases with greater plaque pathology. In double-labeling preparations, BACE1 IR colocalized with immunolabeling for Aβ but not for phosphorylated tau. In perfusion-fixed monkey cortex, locally increased BACE1 IR co-existed with intra-axonal and extracellular Aβ IR among virtually all neuritic plaques, ranging from primitive to typical cored forms. This BACE1 labeling localized to swollen/sprouting axon terminals that might co-express one or another neuronal phenotype markers (GABAergic, glutamatergic, cholinergic, or catecholaminergic). Importantly, these BACE1-labeled dystrophic axons resided near to or in direct contact with blood vessels. These findings suggest that plaque formation in AD or normal aged primates relates to a multisystem axonal pathogenesis that occurs in partnership with a potential vascular or metabolic deficit. The data provide a mechanistic explanation for why senile plaques are present preferentially near the cerebral vasculature. Artículo Texto completo | Immunohistochemistry | | 20726888
|
Beta-secretase-1 elevation in transgenic mouse models of Alzheimer's disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development.
Xue-Mei Zhang,Yan Cai,Kun Xiong,Huaibin Cai,Xue-Gang Luo,Jia-Chun Feng,Richard W Clough,Robert G Struble,Peter R Patrylo,Xiao-Xin Yan
The European journal of neuroscience
30
2009
Mostrar resumen
The presence of neuritic plaques is a pathological hallmark of Alzheimer's disease (AD). However, the origin of extracellular beta-amyloid peptide (Abeta) deposits and the process of plaque development remain poorly understood. The present study attempted to explore plaque pathogenesis by localizing beta-secretase-1 (BACE1) elevation relative to Abeta accumulation and synaptic/neuritic alterations in the forebrain, using transgenic mice harboring familial AD (FAD) mutations (5XFAD and 2XFAD) as models. In animals with fully developed plaque pathology, locally elevated BACE1 immunoreactivity (IR) coexisted with compact-like Abeta deposition, with BACE1 IR occurring selectively in dystrophic axons of various neuronal phenotypes or origins (GABAergic, glutamatergic, cholinergic or catecholaminergic). Prior to plaque onset, localized BACE1/Abeta IR occurred at swollen presynaptic terminals and fine axonal processes. These BACE1/Abeta-containing axonal elements appeared to undergo a continuing process of sprouting/swelling and dystrophy, during which extracellular Abeta IR emerged and accumulated in surrounding extracellular space. These data suggest that BACE1 elevation and associated Abeta overproduction inside the sprouting/dystrophic axonal terminals coincide with the onset and accumulation of extracellular amyloid deposition during the development of neuritic plaques in transgenic models of AD. Our findings appear to be in harmony with an early hypothesis that axonal pathogenesis plays a key or leading role in plaque formation. Artículo Texto completo | | | 20092570
|
Protein 600 is a microtubule/endoplasmic reticulum-associated protein in CNS neurons.
Shim, SY; Wang, J; Asada, N; Neumayer, G; Tran, HC; Ishiguro, K; Sanada, K; Nakatani, Y; Nguyen, MD
The Journal of neuroscience : the official journal of the Society for Neuroscience
28
3604-14
2008
Mostrar resumen
There is an increasing body of literature pointing to cytoskeletal proteins as spatial organizers and interactors of organelles. In this study, we identified protein 600 (p600) as a novel microtubule-associated protein (MAP) developmentally regulated in neurons. p600 exhibits the unique feature to interact with the endoplasmic reticulum (ER). Silencing of p600 by RNA interference (RNAi) destabilizes neuronal processes in young primary neurons undergoing neurite extension and containing scarce staining of the ER marker Bip. Furthermore, in utero electroporation of p600 RNAi alters neuronal migration, a process that depends on synergistic actions of microtubule dynamics and ER functions. p600-depleted migrating neurons display thin, crooked, and "zigzag" leading process with very few ER membranes. Thus, p600 constitutes the only known MAP to associate with the ER in neurons, and this interaction may impact on multiple cellular processes ranging from neuronal development to neuronal maturation and plasticity. | | | 18385319
|
Postnatal development of the primate hippocampal formation.
Lavenex, P; Banta Lavenex, P; Amaral, DG
Developmental neuroscience
29
179-92
2007
Mostrar resumen
The hippocampal formation is a multicomponent region of the medial temporal lobe preferentially involved in declarative and relational memory processing. Behavioral studies have suggested a protracted functional maturation of these structures in primates, and postnatal developmental abnormalities in the hippocampal formation are thought to contribute to neurodevelopmental disorders, such as autism, schizophrenia, epilepsy and Down syndrome. Despite all that we know about the functional organization of the adult hippocampal formation, notably absent is a systematic study of its postnatal maturation in primates. In this article, we review current knowledge of the structural development of the primate hippocampal formation and present new data on its postnatal neuroanatomical development. We summarize what is known about the neurobiological processes, such as the addition of new neurons, the establishment and elaboration of connectivity, and the neurochemical changes, that underlie the structural development and functional maturation of the primate hippocampal formation. We conclude that there is yet insufficient information to identify distinct developmental windows during which different hippocampal regions undergo specific maturational processes. For this reason, it is currently impossible to determine the ages at which specific hippocampal circuits become structurally mature and potentially capable of supporting defined, age-specific functional processes. Together with work in rodents, systematic studies of the structural development and functional maturation of the monkey hippocampal formation will be necessary to gain insight not only into the types of information processing that it subserves, but also into the specific maturational processes that might be affected in human neurodevelopmental disorders. | | | 17148960
|
Orphan glutamate receptor delta1 subunit required for high-frequency hearing.
Gao, J; Maison, SF; Wu, X; Hirose, K; Jones, SM; Bayazitov, I; Tian, Y; Mittleman, G; Matthews, DB; Zakharenko, SS; Liberman, MC; Zuo, J
Molecular and cellular biology
27
4500-12
2007
Mostrar resumen
The function of the orphan glutamate receptor delta subunits (GluRdelta1 and GluRdelta2) remains unclear. GluRdelta2 is expressed exclusively in the Purkinje cells of the cerebellum, and GluRdelta1 is prominently expressed in inner ear hair cells and neurons of the hippocampus. We found that mice lacking the GluRdelta1 protein displayed significant cochlear threshold shifts for frequencies of greater than 16 kHz. These deficits correlated with a substantial loss of type IV spiral ligament fibrocytes and a significant reduction of endolymphatic potential in high-frequency cochlear regions. Vulnerability to acoustic injury was significantly enhanced; however, the efferent innervation of hair cells and the classic efferent inhibition of outer hair cells were unaffected. Hippocampal and vestibular morphology and function were normal. Our findings show that the orphan GluRdelta1 plays an essential role in high-frequency hearing and ionic homeostasis in the basal cochlea, and the locus encoding GluRdelta1 represents a candidate gene for congenital or acquired high-frequency hearing loss in humans. | | | 17438141
|