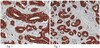
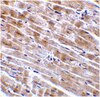
Impaired Ganglioside Metabolism in Huntington's Disease and Neuroprotective Role of GM1.
Maglione V, Marchi P, Di Pardo A, Lingrell S, Horkey M, Tidmarsh E, Sipione S
The Journal of neuroscience : the official journal of the
30
4072-80
2009
Mostrar resumen
Huntington's disease (HD) is a neurodegenerative disorder caused by the expansion of a polyglutamine stretch in the protein huntingtin (Htt). HD neurons are dysfunctional at multiple levels and have increased susceptibility to stress and apoptotic stimuli. We have discovered that synthesis of the ganglioside GM1 is reduced in fibroblasts from HD patients and in cell and animal models of HD, and that decreased GM1 levels contribute to heighten HD cell susceptibility to apoptosis. The apoptotic susceptibility is recapitulated through inhibition of ganglioside synthesis in wild-type striatal cells, suggesting that decreased GM1 levels might be one of the key events leading to HD pathogenesis and progression. Administration of GM1 restores ganglioside levels in HD cells and promotes activation of AKT and phosphorylation of mutant Htt, leading to decreased mutant Htt toxicity and increased survival of HD cells. Our data identify GM1 as a potential treatment for HD. | 20237277
 |
Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model.
Tang, TS; Guo, C; Wang, H; Chen, X; Bezprozvanny, I
The Journal of neuroscience : the official journal of the Society for Neuroscience
29
1257-66
2009
Mostrar resumen
Huntington's disease (HD) is a dominantly inherited, progressive neurodegenerative disease caused by an expanded polyglutamine tract in huntingtin protein (Htt). Medium spiny striatal neurons (MSNs) are primarily affected in HD. Mutant huntingtin protein (Htt(exp)) specifically binds to and activates type 1 inositol 1,4,5-trisphosphate receptor (InsP(3)R1), an intracellular Ca(2+) release channel. Htt(exp)-InsP(3)R1 association is mediated by a cytosolic C-terminal tail of InsP(3)R1 (a 122-aa-long IC10 fragment). To evaluate an importance of Htt(exp) association with InsP(3)R1 for HD pathology, we generated lentiviral and adeno-associated viruses expressing GFP-IC10 fusion protein and performed a series of experiments with YAC128 HD transgenic mouse. Infection with Lenti-GFP-IC10 virus stabilized Ca(2+) signaling in cultured YAC128 MSNs and protected YAC128 MSNs from glutamate-induced apoptosis. Intrastriatal injections of AAV1-GFP-IC10 significantly alleviated motor deficits and reduced MSN loss and shrinkage in YAC128 mice. Our results demonstrate an importance of InsP(3)R1-Htt(exp) association for HD pathogenesis and suggested that InsP(3)R1 is a potential therapeutic target for HD. Our data also support potential use of IC10 peptide as a novel HD therapeutic agent. Artículo Texto completo | 19193873
|
Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington's disease.
Paula A Desplats, James R Lambert, Elizabeth A Thomas, Paula A Desplats, James R Lambert, Elizabeth A Thomas, Paula A Desplats, James R Lambert, Elizabeth A Thomas, Paula A Desplats, James R Lambert, Elizabeth A Thomas
Neurobiology of disease
31
298-308
2008
Mostrar resumen
Transcriptional dysregulation has emerged as a central pathogenic mechanism in Huntington's disease (HD), which is associated with neuropathological changes predominantly in the striatum. Here we demonstrate that expression of Bcl11b (a.k.a. CTIP2), a transcription factor exhibiting highly-enriched localization in adult striatum, is significantly decreased in HD cells, mouse models and human subjects and that overexpression of Bcl11b attenuates toxic effects of mutant huntingtin in cultured striatal neurons. We show that Bcl11b directly activates the proximal promoter regions of striatal-enriched genes and can increase mRNA levels of striatal-expressing genes. We further demonstrate an interaction between Bcl11b and huntingtin protein in cultured cells and brain homogenates from HD R6/1 and YAC72 transgenic mice. We propose that sequestration and/or decreased expression of Bcl11b in HD is responsible, at least in part, for the dysregulation of striatal gene expression observed in HD and may contribute to the specificity of pathology observed in this disease. Artículo Texto completo | 18595722
|
Increased apoptosis, Huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington's disease subjects.
Ciammola, A; Sassone, J; Alberti, L; Meola, G; Mancinelli, E; Russo, MA; Squitieri, F; Silani, V
Cell death and differentiation
13
2068-78
2005
Mostrar resumen
Mutated huntingtin (htt) is ubiquitously expressed in tissues of Huntington's disease (HD) patients. In the brain, the mutated protein leads to neuronal cell dysfunction and death, associated with formation of htt-positive inclusions. Given increasing evidence of abnormalities in HD skeletal muscle, we extensively analyzed primary muscle cell cultures from seven HD subjects (including two unaffected mutation carriers). Myoblasts from presymptomatic and symptomatic HD subjects showed cellular abnormalities in vitro, namely mitochondrial depolarization, cytochrome c release, increased caspase-3, -8, and -9 activities, and defective cell differentiation. Another notable feature was the formation of htt inclusions in differentiated myotubes. This study helps to advance current knowledge about the downstream effects of the htt mutation in human tissues. Further applications may include drug screening using this human cellular model. | 16729030
|
Huntingtin distribution among striatal output neurons of normal rat brain
Fusco, F.R. et al.
Neuroscience Letters , 339:53-56 (2003)
2003
| 12618299
|
Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription.
Kegel, KB; Meloni, AR; Yi, Y; Kim, YJ; Doyle, E; Cuiffo, BG; Sapp, E; Wang, Y; Qin, ZH; Chen, JD; Nevins, JR; Aronin, N; DiFiglia, M
The Journal of biological chemistry
277
7466-76
2002
Mostrar resumen
Huntingtin is a protein of unknown function that contains a polyglutamine tract, which is expanded in patients with Huntington's disease (HD). We investigated the localization and a potential function for huntingtin in the nucleus. In human fibroblasts from normal and HD patients, huntingtin localized diffusely in the nucleus and in subnuclear compartments identified as speckles, promyelocytic leukemia protein bodies, and nucleoli. Huntingtin-positive nuclear bodies redistributed after treatment with sodium butyrate. By Western blot, purified nuclei had low levels of full-length huntingtin compared with the cytoplasm but contained high levels of N- and C-terminal huntingtin fragments, which tightly bound the nuclear matrix. Full-length huntingtin co-immunoprecipitated with the transcriptional corepressor C-terminal binding protein, and polyglutamine expansion in huntingtin reduced this interaction. Full-length wild-type and mutant huntingtin repressed transcription when targeted to DNA. Truncated N-terminal mutant huntingtin repressed transcription, whereas the corresponding wild-type fragment did not repress transcription. We speculate that wild-type huntingtin may function in the nucleus in the assembly of nuclear matrix-bound protein complexes involved with transcriptional repression and RNA processing. Proteolysis of mutant huntingtin may alter nuclear functions by disrupting protein complexes and inappropriately repressing transcription in HD. | 11739372
|
Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease.
F R Fusco, Q Chen, W J Lamoreaux, G Figueredo-Cardenas, Y Jiao, J A Coffman, D J Surmeier, M G Honig, L R Carlock, A Reiner
The Journal of neuroscience : the official journal of the Society for Neuroscience
19
1189-202
1998
Mostrar resumen
Immunohistochemistry and single-cell RT-PCR were used to characterize the localization of huntingtin and/or its mRNA in the major types of striatal neurons and in corticostriatal projection neurons in rats. Single-label immunohistochemical studies revealed that striatum contains scattered large neurons rich in huntingtin and more numerous medium-sized neurons moderate in huntingtin. Double-label immunohistochemical studies showed that the large huntingtin-rich striatal neurons include nearly all cholinergic interneurons and some parvalbuminergic interneurons. Somatostatinergic striatal interneurons, which are medium in size, rarely contained huntingtin. Calbindin immunolabeling showed that the vast majority of the medium-sized striatal neurons that contain huntingtin are projection neurons, but only approximately 65% of calbindin-labeled projection neurons (localized to the matrix compartment of striatum) were labeled for huntingtin. Calbindin-containing projection neurons of the matrix compartment and calbindin-negative projection neurons of the striatal patch compartment contained huntingtin with comparable frequency. Single-cell RT-PCR confirmed that striatal cholinergic interneurons contain huntingtin, but only approximately 65% of projection neurons contained detectable huntingtin message. The finding that huntingtin is not consistently found in striatal projection neurons [which die in Huntington's disease (HD)] but is abundant in striatal cholinergic interneurons (which survive in Huntington's disease) suggests that the mutation in huntingtin that causes HD may not directly kill neurons. In contrast to the heterogeneous expression of huntingtin in the different striatal neuron types, we found all corticostriatal neurons to be rich in huntingtin protein and mRNA. One possibility raised by our findings is that the HD mutation may render corticostriatal neurons destructive rather than render striatal neurons vulnerable. | 9952397
|
Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form.
Trottier, Y, et al.
Nat. Genet., 10: 104-10 (1995)
1994
Mostrar resumen
Huntington's disease (HD) results from the expansion of a polyglutamine encoding CAG repeat in a gene of unknown function. The wide expression of this transcript does not correlate with the pattern of neuropathology in HD. To study the HD gene product (huntingtin), we have developed monoclonal antibodies raised against four different regions of the protein. On western blots, these monoclonals detect the approximately 350 kD huntingtin protein in various human cell lines and in neural and non-neural rodent tissues. In cell lines from HD patients, a doublet protein is detected corresponding to the mutated and normal huntingtin. Immunohistochemical studies in the human brain using two of these antibodies detects the huntingtin in perikarya of some neurons, neuropiles, varicosities and as punctate staining likely to be nerve endings. | 7647777
|