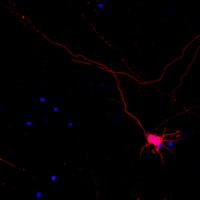
Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination.
Linden, JR; Ma, Y; Zhao, B; Harris, JM; Rumah, KR; Schaeren-Wiemers, N; Vartanian, T
mBio
6
e02513
2015
Abstract anzeigen
Clostridium perfringens epsilon toxin (ε-toxin) is responsible for a devastating multifocal central nervous system (CNS) white matter disease in ruminant animals. The mechanism by which ε-toxin causes white matter damage is poorly understood. In this study, we sought to determine the molecular and cellular mechanisms by which ε-toxin causes pathological changes to white matter. In primary CNS cultures, ε-toxin binds to and kills oligodendrocytes but not astrocytes, microglia, or neurons. In cerebellar organotypic culture, ε-toxin induces demyelination, which occurs in a time- and dose-dependent manner, while preserving neurons, astrocytes, and microglia. ε-Toxin specificity for oligodendrocytes was confirmed using enriched glial culture. Sensitivity to ε-toxin is developmentally regulated, as only mature oligodendrocytes are susceptible to ε-toxin; oligodendrocyte progenitor cells are not. ε-Toxin sensitivity is also dependent on oligodendrocyte expression of the proteolipid myelin and lymphocyte protein (MAL), as MAL-deficient oligodendrocytes are insensitive to ε-toxin. In addition, ε-toxin binding to white matter follows the spatial and temporal pattern of MAL expression. A neutralizing antibody against ε-toxin inhibits oligodendrocyte death and demyelination. This study provides several novel insights into the action of ε-toxin in the CNS. (i) ε-Toxin causes selective oligodendrocyte death while preserving all other neural elements. (ii) ε-Toxin-mediated oligodendrocyte death is a cell autonomous effect. (iii) The effects of ε-toxin on the oligodendrocyte lineage are restricted to mature oligodendrocytes. (iv) Expression of the developmentally regulated proteolipid MAL is required for the cytotoxic effects. (v) The cytotoxic effects of ε-toxin can be abrogated by an ε-toxin neutralizing antibody.Our intestinal tract is host to trillions of microorganisms that play an essential role in health and homeostasis. Disruption of this symbiotic relationship has been implicated in influencing or causing disease in distant organ systems such as the brain. Epsilon toxin (ε-toxin)-carrying Clostridium perfringens strains are responsible for a devastating white matter disease in ruminant animals that shares similar features with human multiple sclerosis. In this report, we define the mechanism by which ε-toxin causes white matter disease. We find that ε-toxin specifically targets the myelin-forming cells of the central nervous system (CNS), oligodendrocytes, leading to cell death. The selectivity of ε-toxin for oligodendrocytes is remarkable, as other cells of the CNS are unaffected. Importantly, ε-toxin-induced oligodendrocyte death results in demyelination and is dependent on expression of myelin and lymphocyte protein (MAL). These results help complete the mechanistic pathway from bacteria to brain by explaining the specific cellular target of ε-toxin within the CNS. | | | 26081637
 |
When larger brains do not have more neurons: increased numbers of cells are compensated by decreased average cell size across mouse individuals.
Herculano-Houzel, S; Messeder, DJ; Fonseca-Azevedo, K; Pantoja, NA
Frontiers in neuroanatomy
9
64
2015
Abstract anzeigen
There is a strong trend toward increased brain size in mammalian evolution, with larger brains composed of more and larger neurons than smaller brains across species within each mammalian order. Does the evolution of increased numbers of brain neurons, and thus larger brain size, occur simply through the selection of individuals with more and larger neurons, and thus larger brains, within a population? That is, do individuals with larger brains also have more, and larger, neurons than individuals with smaller brains, such that allometric relationships across species are simply an extension of intraspecific scaling? Here we show that this is not the case across adult male mice of a similar age. Rather, increased numbers of neurons across individuals are accompanied by increased numbers of other cells and smaller average cell size of both types, in a trade-off that explains how increased brain mass does not necessarily ensue. Fundamental regulatory mechanisms thus must exist that tie numbers of neurons to numbers of other cells and to average cell size within individual brains. Finally, our results indicate that changes in brain size in evolution are not an extension of individual variation in numbers of neurons, but rather occur through step changes that must simultaneously increase numbers of neurons and cause cell size to increase, rather than decrease. | | | 26082686
|
Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling.
Zuckermann, M; Hovestadt, V; Knobbe-Thomsen, CB; Zapatka, M; Northcott, PA; Schramm, K; Belic, J; Jones, DT; Tschida, B; Moriarity, B; Largaespada, D; Roussel, MF; Korshunov, A; Reifenberger, G; Pfister, SM; Lichter, P; Kawauchi, D; Gronych, J
Nature communications
6
7391
2015
Abstract anzeigen
In vivo functional investigation of oncogenes using somatic gene transfer has been successfully exploited to validate their role in tumorigenesis. For tumour suppressor genes this has proven more challenging due to technical aspects. To provide a flexible and effective method for investigating somatic loss-of-function alterations and their influence on tumorigenesis, we have established CRISPR/Cas9-mediated somatic gene disruption, allowing for in vivo targeting of TSGs. Here we demonstrate the utility of this approach by deleting single (Ptch1) or multiple genes (Trp53, Pten, Nf1) in the mouse brain, resulting in the development of medulloblastoma and glioblastoma, respectively. Using whole-genome sequencing (WGS) we characterized the medulloblastoma-driving Ptch1 deletions in detail and show that no off-targets were detected in these tumours. This method provides a fast and convenient system for validating the emerging wealth of novel candidate tumour suppressor genes and the generation of faithful animal models of human cancer. | | | 26067104
|
Pre-existing astrocytes form functional perisynaptic processes on neurons generated in the adult hippocampus.
Krzisch, M; Temprana, SG; Mongiat, LA; Armida, J; Schmutz, V; Virtanen, MA; Kocher-Braissant, J; Kraftsik, R; Vutskits, L; Conzelmann, KK; Bergami, M; Gage, FH; Schinder, AF; Toni, N
Brain structure & function
220
2027-42
2015
Abstract anzeigen
The adult dentate gyrus produces new neurons that morphologically and functionally integrate into the hippocampal network. In the adult brain, most excitatory synapses are ensheathed by astrocytic perisynaptic processes that regulate synaptic structure and function. However, these processes are formed during embryonic or early postnatal development and it is unknown whether astrocytes can also ensheathe synapses of neurons born during adulthood and, if so, whether they play a role in their synaptic transmission. Here, we used a combination of serial-section immuno-electron microscopy, confocal microscopy, and electrophysiology to examine the formation of perisynaptic processes on adult-born neurons. We found that the afferent and efferent synapses of newborn neurons are ensheathed by astrocytic processes, irrespective of the age of the neurons or the size of their synapses. The quantification of gliogenesis and the distribution of astrocytic processes on synapses formed by adult-born neurons suggest that the majority of these processes are recruited from pre-existing astrocytes. Furthermore, the inhibition of astrocytic glutamate re-uptake significantly reduced postsynaptic currents and increased paired-pulse facilitation in adult-born neurons, suggesting that perisynaptic processes modulate synaptic transmission on these cells. Finally, some processes were found intercalated between newly formed dendritic spines and potential presynaptic partners, suggesting that they may also play a structural role in the connectivity of new spines. Together, these results indicate that pre-existing astrocytes remodel their processes to ensheathe synapses of adult-born neurons and participate to the functional and structural integration of these cells into the hippocampal network. | | | 24748560
|
Nicotine accelerates diabetes-induced retinal changes.
Boretsky, A; Gupta, P; Tirgan, N; Liu, R; Godley, BF; Zhang, W; Tilton, RG; Motamedi, M
Current eye research
40
368-77
2015
Abstract anzeigen
To investigate the effects of nicotine on retinal alterations in early-stage diabetes in an established rodent model.Sprague-Dawley rats were examined using a combination of confocal scanning laser ophthalmoscopy and spectral domain optical coherence tomography to determine changes in retinal structure in response to nicotine exposure, diabetes and the combined effects of nicotine and diabetes. Diabetes was induced by a single injection of 65 mg/kg streptozotocin and nicotine injections were administered subcutaneously daily. Retinal thickness in the superior, inferior, nasal and temporal quadrants were determined based on the spectral domain optical coherence tomography (SD-OCT) volume scans (20° × 20°) centered on the optic disc. Segmentation of discrete retinal layers was performed on a subset of SD-OCT cross-sections to further examine changes in each treatment group. Survival of neurons within the ganglion cell layer (GCL) was assessed by confocal morphometric imaging.The control group did not experience any significant change throughout the study. The nicotine treatment group experienced an average decrease in total retinal thickness (TRT) of 9.4 µm with the majority of the loss localized within the outer nuclear layer (ONL) as determined by segmentation analysis (p less than 0.05). The diabetic group exhibited a trend toward decreased TRT while segmentation analysis of the diabetic retinopathy (DR) group revealed significant thinning within the ONL (p less than 0.05). The combination of nicotine and diabetes revealed a significant increase of 8.9 µm in the TRT (p less than 0.05) accompanied by a decrease in the number of GCL neurons.We demonstrated significant temporal changes in retinal morphology in response to nicotine exposure, diabetes and with the combined effects of nicotine and diabetes. These findings may have implications in determining treatment strategies for diabetic patients using products containing nicotine, such as cigarettes, smokeless tobacco, electronic cigarettes or smoking cessation products. | | | 24911405
|
An anterograde rabies virus vector for high-resolution large-scale reconstruction of 3D neuron morphology.
Haberl, MG; Viana da Silva, S; Guest, JM; Ginger, M; Ghanem, A; Mulle, C; Oberlaender, M; Conzelmann, KK; Frick, A
Brain structure & function
220
1369-79
2015
Abstract anzeigen
Glycoprotein-deleted rabies virus (RABV ∆G) is a powerful tool for the analysis of neural circuits. Here, we demonstrate the utility of an anterograde RABV ∆G variant for novel neuroanatomical approaches involving either bulk or sparse neuronal populations. This technology exploits the unique features of RABV ∆G vectors, namely autonomous, rapid high-level expression of transgenes, and limited cytotoxicity. Our vector permits the unambiguous long-range and fine-scale tracing of the entire axonal arbor of individual neurons throughout the brain. Notably, this level of labeling can be achieved following infection with a single viral particle. The vector is effective over a range of ages (greater than 14 months) aiding the studies of neurodegenerative disorders or aging, and infects numerous cell types in all brain regions tested. Lastly, it can also be readily combined with retrograde RABV ∆G variants. Together with other modern technologies, this tool provides new possibilities for the investigation of the anatomy and physiology of neural circuits. | | | 24723034
|
Enrichment rescues contextual discrimination deficit associated with immediate shock.
Clemenson, GD; Lee, SW; Deng, W; Barrera, VR; Iwamoto, KS; Fanselow, MS; Gage, FH
Hippocampus
25
385-92
2015
Abstract anzeigen
Adult animals continue to modify their behavior throughout life, a process that is highly influenced by past experiences. To shape behavior, specific mechanisms of neural plasticity to learn, remember, and recall information are required. One of the most robust examples of adult plasticity in the brain occurs in the dentate gyrus (DG) of the hippocampus, through the process of adult neurogenesis. Adult neurogenesis is strongly upregulated by external factors such as voluntary wheel running (RUN) and environmental enrichment (EE); however, the functional differences between these two factors remain unclear. Although both manipulations result in increased neurogenesis, RUN dramatically increases the proliferation of newborn cells and EE promotes their survival. We hypothesize that the method by which these newborn neurons are induced influences their functional role. Furthermore, we examine how EE-induced neurons may be primed to encode and recognize features of novel environments due to their previous enrichment experience. Here, we gave mice a challenging contextual fear-conditioning (FC) procedure to tease out the behavioral differences between RUN-induced neurogenesis and EE-induced neurogenesis. Despite the robust increases in neurogenesis seen in the RUN mice, we found that only EE mice were able to discriminate between similar contexts in this task, indicating that EE mice might use a different cognitive strategy when processing contextual information. Furthermore, we showed that this improvement was dependent on EE-induced neurogenesis, suggesting a fundamental functional difference between RUN-induced neurogenesis and EE-induced neurogenesis. | | | 25330953
|
The expression of Mas-receptor of the renin-angiotensin system in the human eye.
Vaajanen, A; Kalesnykas, G; Vapaatalo, H; Uusitalo, H
Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv für klinische und experimentelle Ophthalmologie
253
1053-9
2015
Abstract anzeigen
The local renin-angiotensin system has been held to be expressed in many organs, including the eye. It has an important role in the regulation of local fluid homeostasis, cell proliferation, fibrosis, and vascular tone. Mas-receptor (Mas-R) is a potential receptor acting mainly opposite to the well-known angiotensin II receptor type 1. The aim of this study was to determine if Mas-R is expressed in the human eye.Seven enucleated human eyes were used in immunohistochemical detection of Mas-R and its endogenous ligand angiotensin (1-7) [Ang(1-7)]. Both light microscopy and immunofluorescent detection methods were used. A human kidney preparation sample was used as control.The Mas-R was found to have nuclear localization, and localized in the retinal nuclear layers and in the structures of the anterior segment of the eye. A cytoplasmic immunostaining pattern of Ang(1-7) was found in the inner and outer nuclear and plexiform layers of the retina and in the ciliary body.To the best of our knowledge, this is the first report showing Mas-R expression in the human eye. Its localization suggests that it may have a role in physiological and pathological processes in the anterior part of the eye and in the retina. | | | 25677099
|
Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development.
Quesnel-Vallières, M; Irimia, M; Cordes, SP; Blencowe, BJ
Genes & development
29
746-59
2015
Abstract anzeigen
Alternative splicing (AS) generates vast transcriptomic complexity in the vertebrate nervous system. However, the extent to which trans-acting splicing regulators and their target AS regulatory networks contribute to nervous system development is not well understood. To address these questions, we generated mice lacking the vertebrate- and neural-specific Ser/Arg repeat-related protein of 100 kDa (nSR100/SRRM4). Loss of nSR100 impairs development of the central and peripheral nervous systems in part by disrupting neurite outgrowth, cortical layering in the forebrain, and axon guidance in the corpus callosum. Accompanying these developmental defects are widespread changes in AS that primarily result in shifts to nonneural patterns for different classes of splicing events. The main component of the altered AS program comprises 3- to 27-nucleotide (nt) neural microexons, an emerging class of highly conserved AS events associated with the regulation of protein interaction networks in developing neurons and neurological disorders. Remarkably, inclusion of a 6-nt, nSR100-activated microexon in Unc13b transcripts is sufficient to rescue a neuritogenesis defect in nSR100 mutant primary neurons. These results thus reveal critical in vivo neurodevelopmental functions of nSR100 and further link these functions to a conserved program of neuronal microexon splicing. | | | 25838543
|
Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation.
Elmore, MR; Lee, RJ; West, BL; Green, KN
PloS one
10
e0122912
2015
Abstract anzeigen
Microglia are the primary immune cell in the brain and are postulated to play important roles outside of immunity. Administration of the dual colony-stimulating factor 1 receptor (CSF1R)/c-Kit kinase inhibitor, PLX3397, to adult mice results in the elimination of ~99% of microglia, which remain eliminated for as long as treatment continues. Upon removal of the inhibitor, microglia rapidly repopulate the entire adult brain, stemming from a central nervous system (CNS) resident progenitor cell. Using this method of microglial elimination and repopulation, the role of microglia in both healthy and diseased states can be explored. Here, we examine the responsiveness of newly repopulated microglia to an inflammatory stimulus, as well as determine the impact of these cells on behavior, cognition, and neuroinflammation. Two month-old wild-type mice were placed on either control or PLX3397 diet for 21 d to eliminate microglia. PLX3397 diet was then removed in a subset of animals to allow microglia to repopulate and behavioral testing conducted beginning at 14 d repopulation. Finally, inflammatory profiling of the microglia-repopulated brain in response to lipopolysaccharide (LPS; 0.25 mg/kg) or phosphate buffered saline (PBS) was determined 21 d after inhibitor removal using quantitative real time polymerase chain reaction (RT-PCR), as well as detailed analyses of microglial morphologies. We find mice with repopulated microglia to perform similarly to controls by measures of behavior, cognition, and motor function. Compared to control/resident microglia, repopulated microglia had larger cell bodies and less complex branching in their processes, which resolved over time after inhibitor removal. Inflammatory profiling revealed that the mRNA gene expression of repopulated microglia was similar to normal resident microglia and that these new cells appear functional and responsive to LPS. Overall, these data demonstrate that newly repopulated microglia function similarly to the original resident microglia without any apparent adverse effects in healthy adult mice. | | | 25849463
|