Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model.
Chen, D; Liu, S; Ma, H; Liang, X; Ma, H; Yan, X; Yang, B; Wei, J; Liu, X
Cancer cell international
15
42
2015
显示摘要
Mesenchymal stem cells (MSCs) in tumors have emerged as progenitors involved in stroma formation and metastasis of cancers, partially owing to their abilities to differentially express paracrine factors related to the proliferation and invasion of cancer cells. In this regard, increasing evidence has shown that MSCs have impacts on the malignancy of colon cancer, however, the underpinning mechanisms by which MSCs promote cancer metastasis remain elusive.To investigate the crosstalk between adipose-derived MSCs (AMSCs) isolated from adipose tissues and colon cancer cells, a co-culture transwell model of AMSCs and colon cancer cells was employed, and the activation of Wnt signaling and paracrine factors in colon cancer cells and AMSCs were measured.The results showed that AMSCs could enhance the metastatic capacity of colon cancer cells with an elevated expression of mesenchymal-epithelial transition (EMT)-associated genes in a contact-dependent manner. Reciprocally, colon cancer cells were able to induce AMSCs to produce metastasis-related factors and cytokines, such as FGF10, VEGFC and matrix metalloproteinases (MMPs) in part through a mechanism of an activation of Wnt signaling, by which these factors in turn activate Wnt signaling of colon cancer cells. Intriguingly, an inhibition of Wnt signaling leads a reduced capacity of invasion and colony formation of colon cancer cells in vitro, and the tumorigenicity of cancer cells in a murine model.These findings thus suggest that the crosstalk between the Wnt signaling of cancer cells and paracrine factors of AMSCs has an implication in colon cancer malignancy. This study thus uncovers a novel Wnt-paracrine factors mediated-crosstalk between colon cancer cells and AMSCs in cancer malignancy. | | 26060426
 |
PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia.
Radford, H; Moreno, JA; Verity, N; Halliday, M; Mallucci, GR
Acta neuropathologica
130
633-42
2015
显示摘要
The PERK-eIF2α branch of the Unfolded Protein Response (UPR) mediates the transient shutdown of translation in response to rising levels of misfolded proteins in the endoplasmic reticulum. PERK and eIF2α activation are increasingly recognised in postmortem analyses of patients with neurodegenerative disorders, including Alzheimer's disease, the tauopathies and prion disorders. These are all characterised by the accumulation of misfolded disease-specific proteins in the brain in association with specific patterns of neuronal loss, but the role of UPR activation in their pathogenesis is unclear. In prion-diseased mice, overactivation of PERK-P/eIF2α-P signalling results in the sustained reduction in global protein synthesis, leading to synaptic failure, neuronal loss and clinical disease. Critically, restoring vital neuronal protein synthesis rates by inhibiting the PERK-eIF2α pathway, both genetically and pharmacologically, prevents prion neurodegeneration downstream of misfolded prion protein accumulation. Here we show that PERK-eIF2α-mediated translational failure is a key process leading to neuronal loss in a mouse model of frontotemporal dementia, where the misfolded protein is a form of mutant tau. rTg4510 mice, which overexpress the P301L tau mutation, show dysregulated PERK signalling and sustained repression of protein synthesis by 6 months of age, associated with onset of neurodegeneration. Treatment with the PERK inhibitor, GSK2606414, from this time point in mutant tau-expressing mice restores protein synthesis rates, protecting against further neuronal loss, reducing brain atrophy and abrogating the appearance of clinical signs. Further, we show that PERK-eIF2α activation also contributes to the pathological phosphorylation of tau in rTg4510 mice, and that levels of phospho-tau are lowered by PERK inhibitor treatment, providing a second mechanism of protection. The data support UPR-mediated translational failure as a generic pathogenic mechanism in protein-misfolding disorders, including tauopathies, that can be successfully targeted for prevention of neurodegeneration. | | 26450683
|
Time-dependent activation of MAPK/Erk1/2 and Akt/GSK3 cascades: modulation by agomelatine.
Musazzi, L; Seguini, M; Mallei, A; Treccani, G; Pelizzari, M; Tornese, P; Racagni, G; Tardito, D
BMC neuroscience
15
119
2014
显示摘要
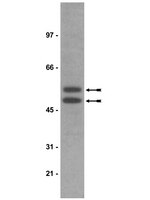
The novel antidepressant agomelatine, a melatonergic MT1/MT2 agonist combined with 5-HT2c serotonin antagonist properties, showed antidepressant action in preclinical and clinical studies. There is a general agreement that the therapeutic action of antidepressants needs the activation of slow-onset adaptations in downstream signalling pathways finally regulating neuroplasticity. In the last several years, particular attention was given to cAMP-responsive element binding protein (CREB)-related pathways, since it was shown that chronic antidepressants increase CREB phosphorylation and transcriptional activity, through the activation of calcium/calmodulin-dependent (CaM) and mitogen activated protein kinase cascades (MAPK/Erk1/2). Aim of this work was to analyse possible effects of chronic agomelatine on time-dependent changes of different intracellular signalling pathways in hippocampus and prefrontal/frontal cortex of male rats. To this end, measurements were performed 1 h or 16 h after the last agomelatine or vehicle injection.We have found that in naïve rats chronic agomelatine, contrary to traditional antidepressants, did not increase CREB phosphorylation, but modulates the time-dependent regulation of MAPK/Erk1/2 and Akt/glycogen synthase kinase-3 (GSK-3) pathways.Our results suggest that the intracellular molecular mechanisms modulated by chronic agomelatine may be partly different from those of traditional antidepressants and involve the time-dependent regulation of MAPK/Erk1/2 and Akt/GSK-3 signalling pathways. This could exert a role in the antidepressant efficacy of the drug. | Western Blotting | 25332063
|
Increased Tau Phosphorylation and Impaired Brain Insulin/IGF Signaling in Mice Fed a High Fat/High Cholesterol Diet.
Bhat, Narayan R and Thirumangalakudi, Lakshmi
J. Alzheimers Dis., (2013)
2013
显示摘要
Previous studies demonstrated that a high fat/high cholesterol (HFC) diet results in a loss of working memory in mice correlated with neuroinflammatory changes and increased AβPP processing (Thirumangalakudi et al. (2008) J Neurochem 106, 475-485). To further explore the nature of the molecular correlates of cognitive impairment, in this study, we examined changes in tau phosphorylation, insulin/IGF-1 signaling (IIS) including GSK3, and levels of specific synaptic proteins. Immunoblot analysis of hippocampal tissue from C57BL/6 mice fed HFC for 2 months with anti-phospho-tau (i.e., PHF1 and phospho-Thr-231 tau) antibodies demonstrated the presence of hyperphosphorylated tau. The tau phosphorylation correlated with activated GSK3, a prominent tau kinase normally kept inactive under the control of IIS. That IIS itself was impaired due to the hyperlipidemic diet was confirmed by a down-regulation of insulin receptor substrate-1 and phospho-Akt and levels. Although no significant changes in the levels of the pre-synaptic protein (i.e., synaptophysin) in response to HFC were apparent in immunoblot analysis, there was a clear down-regulation of the post-synaptic protein, PSD95, and drebrin, a dendritic spine-specific protein, indicative of altered synaptic plasticity. The results, in concert with previous findings with the same model, suggest that high dietary fat/cholesterol elicits brain insulin resistance and altered IIS leading to Alzheimer's disease-like cognitive impairment in 'normal' mice. | | 23703152
|
Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma.
Biechele, TL; Kulikauskas, RM; Toroni, RA; Lucero, OM; Swift, RD; James, RG; Robin, NC; Dawson, DW; Moon, RT; Chien, AJ
Science signaling
5
ra3
2012
显示摘要
Because the Wnt/β-catenin signaling pathway is linked to melanoma pathogenesis and to patient survival, we conducted a kinome small interfering RNA (siRNA) screen in melanoma cells to expand our understanding of the kinases that regulate this pathway. We found that BRAF signaling, which is constitutively activated in many melanomas by the BRAF(V600E) mutation, inhibits Wnt/β-catenin signaling in human melanoma cells. Because inhibitors of BRAF(V600E) show promise in ongoing clinical trials, we investigated whether altering Wnt/β-catenin signaling might enhance the efficacy of the BRAF(V600E) inhibitor PLX4720. We found that endogenous β-catenin was required for PLX4720-induced apoptosis of melanoma cells and that activation of Wnt/β-catenin signaling synergized with PLX4720 to decrease tumor growth in vivo and to increase apoptosis in vitro. This synergistic enhancement of apoptosis correlated with reduced abundance of an endogenous negative regulator of β-catenin, AXIN1. In support of the hypothesis that AXIN1 is a mediator rather than a marker of apoptosis, siRNA directed against AXIN1 rendered resistant melanoma cell lines susceptible to apoptosis in response to treatment with a BRAF(V600E) inhibitor. Thus, Wnt/β-catenin signaling and AXIN1 may regulate the efficacy of inhibitors of BRAF(V600E), suggesting that manipulation of the Wnt/β-catenin pathway could be combined with BRAF inhibitors to treat melanoma. | | 22234612
|
Inhibition of cyclin-dependent kinase 5 but not of glycogen synthase kinase 3-β prevents neurite retraction and tau hyperphosphorylation caused by secretable products of human T-cell leukemia virus type I-infected lymphocytes.
Maldonado, H; Ramírez, E; Utreras, E; Pando, ME; Kettlun, AM; Chiong, M; Kulkarni, AB; Collados, L; Puente, J; Cartier, L; Valenzuela, MA
Journal of neuroscience research
89
1489-98
2011
显示摘要
Human T-cell leukemia virus type I (HTLV-I)-associated myelopathy/tropical spastic paraparesis (HAM/TSP) is a neurodegenerative disease characterized by selective loss of axons and myelin in the corticospinal tracts. This central axonopathy may originate from the impairment of anterograde axoplasmic transport. Previous work showed tau hyperphosphorylation at T(181) in cerebrospinal fluid of HAM/TSP patients. Similar hyperphosphorylation occurs in SH-SY5Y cells incubated with supernatant from MT-2 cells (HTLV-I-infected lymphocytes secreting viral proteins, including Tax) that produce neurite shortening. Tau phosphorylation at T(181) is attributable to glycogen synthase kinase 3-β (GSK3-β) and cyclin-dependent kinase 5 (CDK5) activation. Here we investigate whether neurite retraction in the SH-SY5Y model associates with concurrent changes in other tau hyperphosphorylable residues. Threonine 181 turned out to be the only tau hyperphosphorylated residue. We also evaluate the role of GSK3-β and CDK5 in this process by using specific kinase inhibitors (LiCl, TDZD-8, and roscovitine). Changes in both GSK3-β active and inactive forms were followed by measuring the regulatory phosphorylable sites (S(9) and Y(216) , inactivating and activating phosphorylation, respectively) together with changes in β-catenin protein levels. Our results showed that LiCl and TDZD-8 were unable to prevent MT-2 supernatant-mediated neurite retraction and also that neither Y(216) nor S(9) phosphorylations were changed in GSK3-β. Thus, GSK3-β seems not to play a role in T(181) hyperphosphorylation. On the other hand, the CDK5 involvement in tau phosphorylation was confirmed by both the increase in its enzymatic activity and the absence of MT-2 neurite retraction in the presence of roscovitine or CDK5 siRNA transfection. | | 21671254
|
Compression alters kinase and phosphatase activity and tau and MAP2 phosphorylation transiently while inducing the fast adaptive dendritic remodeling of underlying cortical neurons.
Li-Jin Chen,Yueh-Jan Wang,Guo-Fang Tseng
Journal of neurotrauma
27
2010
显示摘要
In traumatic brain injury (TBI) there is often compression of the cerebral cortex. Using a rat epidural bead implantation model we found that mechanical compression distorted the dendrites of underlying cortical pyramidal neurons, and that the deformed dendrites regained straight morphology in 3 days. This was accompanied by a transient increase in the phosphorylation of microtubule-associated proteins (MAPs) at sites known to destabilize microtubules, including MAP2 from 30 min to 1 h, and tau from 10 min to 12 h following compression. Immunostaining confirmed that phosphorylated MAPs were concentrated at the somata and dendrites of compressed cortical pyramidal neurons. Enzymes regulating MAP phosphorylation were found to be simultaneously altered, including downregulation of protein phosphatase 2A, but not 2B, activity from 10 min to 1 day, and transient excitatory phosphorylation of extracellular signal-regulated protein kinase 1/2 and p38/mitogen-activated protein kinase. The temporal coincidence of these events suggests that alterations of phosphatase and kinase activity underlie MAP2 and tau phosphorylation, thus causing the compressed cortical neurons to remodel their dendrites, including the proximal segments. The rapid onset of these molecular changes demonstrates that compression causes cortical neurons to undergo active changes much early than expected. The large-scale structural changes that result can alter cortical function for an extended period of time. | | 20568963
|
Activation of canonical wingless-type MMTV integration site family (Wnt) signaling in mature adipocytes increases beta-catenin levels and leads to cell dedifferentiation and insulin resistance.
Gustafson, B; Smith, U
The Journal of biological chemistry
285
14031-41
2010
显示摘要
Canonical Wnt ligands are secreted by several cell types in the adipose tissue. We examined if mature adipocytes can also be target cells and found that canonical Wnt activation by Wnt3a induced a marked dedifferentiation of both 3T3-L1 and human adipocytes. Typical adipogenic markers were reduced while undifferentiated cell markers like Pref-1/Dlk1, Wnt10b, and Gata2 were increased. The cells also became insulin-resistant with impaired upstream insulin signaling and reduced glucose uptake. Wnt3a stabilized beta-catenin in the absence of the LRP6 receptor and with maintained axin and Dickkopf-1 protein expression. PPARgamma was repressed and PPARgamma ligands could not restore the adipogenic markers or reduce the beta-catenin levels. The dedifferentiated adipocytes expressed the myofibroblast marker alpha-smooth muscle actin and were also susceptible to osteogenic transdifferentiation. These results identify a novel pathway in mature adipose cells that is critical for maintaining the normal adipocyte phenotype and insulin sensitivity. | Western Blotting | 20179324
|
Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration.
Varrin-Doyer, M; Vincent, P; Cavagna, S; Auvergnon, N; Noraz, N; Rogemond, V; Honnorat, J; Moradi-Améli, M; Giraudon, P
The Journal of biological chemistry
284
13265-76
2009
显示摘要
In the central nervous system, collapsin response mediator protein 2 (CRMP2) is a transducer protein that supports the semaphorin-induced guidance of axons toward their cognate target. However, we previously showed that CRMP2 is also expressed in immune cells and plays a crucial role in T lymphocyte migration. Here we further investigated the molecular mechanisms underlying CRMP2 function in chemokine-directed T-cell motility. Examining Jurkat T-cells treated with the chemokine CXCL12, we found that 1) CXCL12 induces a dynamic re-localization of CRMP2 to uropod, the flexible structure of migrating lymphocyte, and increases its binding to the cytoskeletal protein vimentin; 2) CXCL12 decreases phosphorylation of the glycogen synthase kinase-3beta-targeted residues CRMP2-Thr-509/514; and 3) tyrosine Tyr-479 is a new phosphorylation CRMP2 residue and a target for the Src-family kinase Yes. Moreover, phospho-Tyr-479 increased under CXCL12 signaling while phospho-Thr-509/514 decreased. The functional importance of this tyrosine phosphorylation was demonstrated by Y479F mutation that strongly reduced CXCL12-mediated T-cell polarization and motility as tested in a transmigration model and on neural tissue. We propose that differential phosphorylation by glycogen synthase kinase-3beta and Yes modulates the contribution of CRMP2 to cytoskeletal reorganization during chemokine-directed T-cell migration. In addition to providing a novel mechanism for T lymphocyte motility, our findings reveal CRMP2 as a transducer of chemokine signaling. 全文本文章 | | 19276087
|
Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation.
M S Song, G Rauw, G B Baker, S Kar, M S Song, G Rauw, G B Baker, S Kar
The European journal of neuroscience
28
1989-2002
2008
显示摘要
It has been suggested that accumulation of beta-amyloid (Abeta) peptide triggers neurodegeneration, at least in part, via glutamate-mediated excitotoxicity in Alzheimer's disease (AD) brain. This is supported by observations that toxicity induced by Abeta peptide in cultured neurons and in adult rat brain is known to be mediated by activation of glutamatergic N-methyl-d-aspartate (NMDA) receptors. Additionally, recent clinical studies have shown that memantine, a noncompetitive NMDA receptor antagonist, can significantly improve cognitive functions in some AD patients. However, very little is currently known about the potential role of memantine against Abeta-induced toxicity. In the present study, we have shown that Abeta(1-42)-induced toxicity in rat primary cortical cultured neurons is accompanied by increased extracellular and decreased intracellular glutamate levels. We subsequently demonstrated that Abeta toxicity is induced by increased phosphorylation of tau protein and activation of tau kinases, i.e. glycogen synthase kinase-3beta and extracellular signal-related kinase 1/2. Additionally, Abeta treatment induced cleavage of caspase-3 and decreased phosphorylation of cyclic AMP response element binding protein, which are critical in determining survival of neurons. Memantine treatment significantly protected cultured neurons against Abeta-induced toxicity by attenuating tau-phosphorylation and its associated signaling mechanisms. However, this drug did not alter either conformation or internalization of Abeta(1-42) and it was unable to attenuate Abeta-induced potentiation of extracellular glutamate levels. These results, taken together, provide new insights into the possible neuroprotective action of memantine in AD pathology. | | 19046381
|