



05-1532 Sigma-AldrichAnti-SMN Antibody, clone 2B1
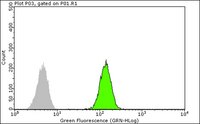


Anti-SMN Antibody, clone 2B1 is a Mouse Monoclonal Antibody for detection of SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease) & has been validated in WB, ICC & IP.
More>> Anti-SMN Antibody, clone 2B1 is a Mouse Monoclonal Antibody for detection of SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease) & has been validated in WB, ICC & IP. Less<<Anti-SMN Antibody, clone 2B1 MSDS (material safety data sheet) or SDS, CoA and CoQ, dossiers, brochures and other available documents.
Recommended Products
概述
| Replacement Information |
|---|
重要规格表
| Species Reactivity | Key Applications | Host | Format | Antibody Type |
|---|---|---|---|---|
| H, M, Xn | WB, ICC, IP | M | Purified | Monoclonal Antibody |
| References |
|---|
| Product Information | |
|---|---|
| Format | Purified |
| Control |
|
| Presentation | Purified mouse monoclonal IgG1κ in buffer containing 0.1 M Tris-Glycine (pH 7.4, 150 mM NaCl) with 0.05% sodium azide. |
| Quality Level | MQ100 |
| Physicochemical Information |
|---|
| Dimensions |
|---|
| Materials Information |
|---|
| Toxicological Information |
|---|
| Safety Information according to GHS |
|---|
| Safety Information |
|---|
| Storage and Shipping Information | |
|---|---|
| Storage Conditions | Stable for 1 year at 2-8°C from date of receipt. |
| Packaging Information | |
|---|---|
| Material Size | 100 µg |
| Transport Information |
|---|
| Supplemental Information |
|---|
| Specifications |
|---|
| Global Trade Item Number | |
|---|---|
| 产品目录编号 | GTIN |
| 05-1532 | 04053252741173 |
Documentation
Anti-SMN Antibody, clone 2B1 MSDS
| 职位 |
|---|
Anti-SMN Antibody, clone 2B1 分析证书
| 标题 | 批号 |
|---|---|
| Anti-SMN, clone 2B1 | 2470958 |
| Anti-SMN, clone 2B1 - 2006960 | 2006960 |
| Anti-SMN, clone 2B1 - 2189741 | 2189741 |
| Anti-SMN, clone 2B1 - 2262736 | 2262736 |
| Anti-SMN, clone 2B1 - 2364201 | 2364201 |
| Anti-SMN, clone 2B1 - 3239899 | 3239899 |
| Anti-SMN, clone 2B1 - 3489750 | 3489750 |
| Anti-SMN, clone 2B1 - 3585808 | 3585808 |
| Anti-SMN, clone 2B1 - 3705804 | 3705804 |
| Anti-SMN, clone 2B1 - 3807942 | 3807942 |
参考
| 参考概述 | 公共医疗ID |
|---|---|
| Autoantibodies to survival of motor neuron complex in patients with polymyositis: immunoprecipitation of D, E, F, and G proteins without other components of small nuclear ribonucleoproteins. Satoh, Minoru, et al. Arthritis Rheum., 63: 1972-8 (2011) 2011 显示摘要 | 21425128
|