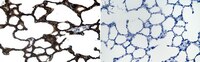
Alveolar type II epithelial cell dysfunction in rat experimental hepatopulmonary syndrome (HPS).
Yang, W; Hu, B; Wu, W; Batra, S; Blackburn, MR; Alcorn, JL; Fallon, MB; Zhang, J
PloS one
9
e113451
2014
显示摘要
The hepatopulmonary syndrome (HPS) develops when pulmonary vasodilatation leads to abnormal gas exchange. However, in human HPS, restrictive ventilatory defects are also observed supporting that the alveolar epithelial compartment may also be affected. Alveolar type II epithelial cells (AT2) play a critical role in maintaining the alveolar compartment by producing four surfactant proteins (SPs, SP-A, SP-B, SP-C and SP-D) which also facilitate alveolar repair following injury. However, no studies have evaluated the alveolar epithelial compartment in experimental HPS. In this study, we evaluated the alveolar epithelial compartment and particularly AT2 cells in experimental HPS induced by common bile duct ligation (CBDL). We found a significant reduction in pulmonary SP production associated with increased apoptosis in AT2 cells after CBDL relative to controls. Lung morphology showed decreased mean alveolar chord length and lung volumes in CBDL animals that were not seen in control models supporting a selective reduction of alveolar airspace. Furthermore, we found that administration of TNF-α, the bile acid, chenodeoxycholic acid, and FXR nuclear receptor activation (GW4064) induced apoptosis and impaired SP-B and SP-C production in alveolar epithelial cells in vitro. These results imply that AT2 cell dysfunction occurs in experimental HPS and is associated with alterations in the alveolar epithelial compartment. Our findings support a novel contributing mechanism in experimental HPS that may be relevant to humans and a potential therapeutic target. | Western Blotting | 25419825
 |
Verification and spatial localization of aquaporin-5 in the ocular lens.
Grey, AC; Walker, KL; Petrova, RS; Han, J; Wilmarth, PA; David, LL; Donaldson, PJ; Schey, KL
Experimental eye research
108
94-102
2013
显示摘要
Until recently, the lens was thought to express only two aquaporin (AQP) water channels, AQP1 and AQP0. In this study we confirm lenticular AQP5 protein expression by Western blotting and mass spectrometry in lenses from a variety of species. In addition, confocal microscopy was used to map cellular distributions of AQP5 in mouse, rat and human lenses. Tandem mass spectrometry of a human lens membrane preparation revealed extensive sequence coverage (56.2%) of AQP5. Western blotting performed on total fiber cell membranes from mouse, rat, bovine and human lenses confirmed AQP5 protein expression is conserved amongst species. Western blotting of dissected lens fractions suggests that AQP5 is processed in the lens core by C-terminal truncation. Immunohistochemistry showed that AQP5 signal was most abundant in the lens outer cortex and decreased in intensity in the lens core. Furthermore, AQP5 undergoes differentiation-dependent changes in subcellular location from an intracellular localization in differentiating fiber cells to the plasma membrane of mature fiber cells upon the loss of fiber cell nuclei. Our results show that AQP5 is a significant component of lens fiber cell membranes, representing the second most abundant water channel in these cells. Together, the changes to AQP5 distribution and structure are likely to modulate the functional role of AQP5 in different regions of the lens. | | 23313152
|
Ephrin-B2 reverse signaling increases α5β1 integrin-mediated fibronectin deposition and reduces distal lung compliance.
Bennett, KM; Afanador, MD; Lal, CV; Xu, H; Persad, E; Legan, SK; Chenaux, G; Dellinger, M; Savani, RC; Dravis, C; Henkemeyer, M; Schwarz, MA
American journal of respiratory cell and molecular biology
49
680-7
2013
显示摘要
Alveolar growth abnormalities and severe respiratory dysfunction are often fatal. Identifying mechanisms that control epithelial proliferation and enlarged, poorly septated airspaces is essential in developing new therapies for lung disease. The membrane-bound ligand ephrin-B2 is strongly expressed in lung epithelium, and yet in contrast to its known requirement for arteriogenesis, considerably less is known regarding the function of this protein in the epithelium. We hypothesize that the vascular mediator ephrin-B2 governs alveolar growth and mechanics beyond the confines of the endothelium. We used the in vivo manipulation of ephrin-B2 reverse signaling to determine the role of this vascular mediator in the pulmonary epithelium and distal lung mechanics. We determined that the ephrin-B2 gene (EfnB2) is strongly expressed in alveolar Type 2 cells throughout development and into adulthood. The role of ephrin-B2 reverse signaling in the lung was assessed in Efnb2(LacZ/6YFΔV) mutants that coexpress the intracellular truncated ephrin-B2-β-galactosidase fusion and an intracellular point mutant ephrin-B2 protein that is unable to become tyrosine-phosphorylated or to interact with either the SH2 or PDZ domain-containing downstream signaling proteins. In these viable mice, we observed pulmonary hypoplasia and altered pulmonary mechanics, as evidenced by a marked reduction in lung compliance. Associated with the reduction in lung compliance was a significant increase in insoluble fibronectin (FN) basement membrane matrix assembly with FN deposition, and a corresponding increase in the α5 integrin receptor required for FN fibrillogenesis. These experiments indicate that ephrin-B2 reverse signaling mediates distal alveolar formation, fibrillogenesis, and pulmonary compliance. | Immunohistochemistry | 23742148
|
An autoimmunized mouse model recapitulates key features in the pathogenesis of Sjögren's syndrome.
Lin, X; Song, JX; Shaw, PC; Ng, TB; Wong, RN; Sze, SC; Tong, Y; Lee, KF; Zhang, KY
International immunology
23
613-24
2011
显示摘要
The pathogenesis of Sjögren's syndrome (SS) is poorly understood. To evaluate an autoimmunization-induced experimental SS model, we firstly observed the phenotype of lymphocyte infiltration in the enlarged submandibular gland (SG). Furthermore, significant activation of caspase-3 and a high ratio of Bax-to-Bcl-2 were detected, indicating the inflammatory apoptosis associated with developmental foci. Meanwhile, the dysregulated cytokines, such as tumor necrosis factor α, IL-1β and IL-6 mRNA expression, were found to be over-expressed. A progressive decrease of aquaporin 5 and its subcellular translocation from apical to basal membrane in SG was found to be associated with the abnormally expressed M3 muscarinic acetylcholine receptor. This pattern was found to be similar to that seen in human SS and possibly contributed to the saliva secretion deficiency. Thus, this autoimmunization-induced model recapitulates the key features of human SS and may have potential for studying the pathogenesis of human SS. | Western Blotting | 21846814
|
Characterization of wild-type and deltaF508 cystic fibrosis transmembrane regulator in human respiratory epithelia.
Kreda, Silvia M, et al.
Mol. Biol. Cell, 16: 2154-67 (2005)
2005
显示摘要
Previous studies in native tissues have produced conflicting data on the localization and metabolic fate of WT and deltaF508 cystic fibrosis transmembrane regulator (CFTR) in the lung. Combining immunocytochemical and biochemical studies utilizing new high-affinity CFTR mAbs with ion transport assays, we examined both 1) the cell type and region specific expression of CFTR in normal airways and 2) the metabolic fate of deltaF508 CFTR and associated ERM proteins in the cystic fibrosis lung. Studies of lungs from a large number of normal subjects revealed that WT CFTR protein localized to the apical membrane of ciliated cells within the superficial epithelium and gland ducts. In contrast, other cell types in the superficial, gland acinar, and alveolar epithelia expressed little WT CFTR protein. No deltaF508 CFTR mature protein or function could be detected in airway specimens freshly excised from a large number of deltaF508 homozygous subjects, despite an intact ERM complex. In sum, our data demonstrate that WT CFTR is predominantly expressed in ciliated cells, and deltaF508 CFTR pathogenesis in native tissues, like heterologous cells, reflects loss of normal protein processing. | | 15716351
|
Expression and localization of epithelial aquaporins in the adult human lung.
Kreda, S M, et al.
Am. J. Respir. Cell Mol. Biol., 24: 224-34 (2001)
2001
显示摘要
Aquaporins (AQPs) facilitate water transport across epithelia and play an important role in normal physiology and disease in the human airways. We used in situ hybridization and immunofluorescence to determine the expression and cellular localization of AQPs 5, 4, and 3 in human airway sections. In nose and bronchial epithelia, AQP5 is expressed at the apical membrane of columnar cells of the superficial epithelium and submucosal gland acinar cells. AQP4 was detected in basolateral membranes in ciliated ducts and by in situ in gland acinar cells. AQP3 is present on basal cells of both superficial epithelium and gland acinus. In these regions AQPs 5, 4, and 3 are appropriately situated to permit transepithelial water permeability. In the small airways (proximal and terminal bronchioles) AQP3 distribution shifts from basal cell to surface expression (i.e., localized to the apical membrane of proximal and terminal bronchioles) and is the only AQP identified in this region of the human lung. The alveolar epithelium has all three AQPs represented, with AQP5 and AQP4 localized to type I pneumocytes and AQP3 to type II cells. This study describes an intricate network of AQP expression that mediates water transport across the human airway epithelium. | | 11245621
|
Cloning and characterization of murine Aqp5: evidence for a conserved aquaporin gene cluster.
Krane, C M, et al.
Mamm. Genome, 10: 498-505 (1999)
1999
显示摘要
Aquaporin 5 (Aqp5), a member of the aquaporin family of membrane water channels, is thought to modulate the osmolality of fluids in the eye, lung, and salivary gland. Here, we report the cloning and genomic characterization of murine Aqp5 and its expression in relevant mouse tissues. This gene, comprised of four exons encoding 265 amino acids (121, 55, 28, and 61 amino acids respectively), is transcribed into an approximate 1.8-kb mRNA detected in lung, parotid, submandibular, sublingual, and lacrimal tissues. Aqp5 encodes a protein that is 98% identical to rat Aqp5. An Aqp5 antibody detects an approximately 27-kDa protein band in mouse lung, and an additional 29 kDa band in salivary gland. Cloning and physical mapping genomic fragments contiguous with Aqp5 revealed two other members of the aquaporin family: Aqp2 and Aqp6, arrayed head to tail in the order Aqp2-Aqp5-Aqp6, and provides evidence of a gene cluster conserved in order and orientation in both mice and humans. | | 10337625
|
The human Aquaporin-5 gene. Molecular characterization and chromosomal localization.
Lee, M D, et al.
J. Biol. Chem., 271: 8599-604 (1996)
1996
显示摘要
The cDNA for the fifth mammalian aquaporin (AQP5) was isolated from rat, and expression was demonstrated in rat salivary and lacrimal glands, cornea, and lung (Raina, S., Preston, G. M., Guggino, W. B., and Agre, P. (1995) J. Biol. Chem. 270, 1908-1912). Here we report the isolation and characterization of the human AQP5 cDNA and gene. The AQP5 cDNA from a human submaxillary gland library contains a 795-base pair open reading frame encoding a 265-amino acid protein. The deduced amino acid sequences of human and rat AQP5 are 91% identical with 6 substitutions in the 22-amino acid COOH-terminal domain. Expression of human AQP5 in Xenopus oocytes conferred mercurial-sensitive osmotic water permeability (Pf) equivalent to other aquaporins. The human AQP5 structural gene resides within a 7. 4-kilobase SalI-EcoRI fragment with four exons corresponding to amino acids 1-121, 122-176, 177-204, and 205-265 separated by introns of 1.2, 0.5, and 0.9 kilobases. A transcription initiation site was identified 518 base pairs upstream of the initiating methionine. Genomic Southern analysis indicated that AQP5 is a single copy gene which localized to human chromosome 12q13; this coincides with the chromosomal locations of the homologous human genes MIP and AQP2, thus confirming 12q13 as the site of an aquaporin gene cluster. The mouse gene localized to distal chromosome 15. This information may permit molecular characterization of AQP5 expression during normal development and in clinical disorders. | | 8621489
|
Molecular cloning and characterization of an aquaporin cDNA from salivary, lacrimal, and respiratory tissues.
Raina, S, et al.
J. Biol. Chem., 270: 1908-12 (1995)
1995
显示摘要
The Aquaporin family of water channels plays a fundamental role in transmembrane water movements in numerous plant and animal tissues. Since the molecular pathway by which water is secreted by salivary glands is unknown, a cDNA was isolated from rat submandibular gland by homology cloning. Similar to other Aquaporins, the salivary cDNA encodes a 265-residue polypeptide with six putative transmembrane domains separated by five connecting loops (A-E); the NH2- and COOH-terminal halves of the polypeptide are sequence-related, and each contains the motif Asn-Pro-Ala. A mercurial-inhibition site is present in extracellular loop E, and cytoplasmic loop D contains a cAMP-protein kinase phosphorylation consensus. In vitro translation yielded a 27-kDa polypeptide, and expression of the cRNA in Xenopus oocytes conferred a 20-fold increase in osmotic water permeability (Pf) which was reversibly inhibited by 1 mM HgCl2. Northern analysis demonstrated a 1.6-kilobase mRNA in submandibular, parotid, and sublingual salivary glands, lacrimal gland, eye, trachea, and lung. In situ hybridization revealed a strong hybridization over the corneal epithelium in eye and over the secretory lobules in salivary glands. These studies have identified a new mammalian member of the Aquaporin water channel family (gene symbol AQP5) which is implicated in the generation of saliva, tears, and pulmonary secretions. | | 7530250
|