Epinephrine Activation of the β2-Adrenoceptor Is Required for IL-13-Induced Mucin Production in Human Bronchial Epithelial Cells.
Al-Sawalha, N; Pokkunuri, I; Omoluabi, O; Kim, H; Thanawala, VJ; Hernandez, A; Bond, RA; Knoll, BJ
PloS one
10
e0132559
2015
Mostrar resumen
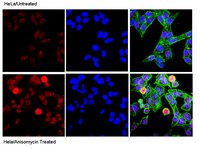
Mucus hypersecretion by airway epithelium is a hallmark of inflammation in allergic asthma and results in airway narrowing and obstruction. Others have shown that administration a TH2 cytokine, IL-13 is sufficient to cause mucus hypersecretion in vivo and in vitro. Asthma therapy often utilizes β2-adrenoceptor (β2AR) agonists, which are effective acutely as bronchodilators, however chronic use may lead to a worsening of asthma symptoms. In this study, we asked whether β2AR signaling in normal human airway epithelial (NHBE) cells affected mucin production in response to IL-13. This cytokine markedly increased mucin production, but only in the presence of epinephrine. Mucin production was blocked by ICI-118,551, a preferential β2AR antagonist, but not by CGP-20712A, a preferential β1AR antagonist. Constitutive β2AR activity was not sufficient for IL-13 induced mucin production and β-agonist-induced signaling is required. A clinically important long-acting β-agonist, formoterol, was as effective as epinephrine in potentiating IL-13 induced MUC5AC transcription. IL-13 induced mucin production in the presence of epinephrine was significantly reduced by treatment with selective inhibitors of ERK1/2 (FR180204), p38 (SB203580) and JNK (SP600125). Replacement of epinephrine with forskolin + IBMX resulted in a marked increase in mucin production in NHBE cells in response to IL-13, and treatment with the inhibitory cAMP analogue Rp-cAMPS decreased mucin levels induced by epinephrine + IL-13. Our findings suggest that β2AR signaling is required for mucin production in response to IL-13, and that mitogen activated protein kinases and cAMP are necessary for this effect. These data lend support to the notion that β2AR-agonists may contribute to asthma exacerbations by increasing mucin production via activation of β2ARs on epithelial cells. | | | 26161982
 |
RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38.
Weilbacher, A; Gutekunst, M; Oren, M; Aulitzky, WE; van der Kuip, H
Cell death & disease
5
e1318
2014
Mostrar resumen
Significant advances have been made in the development of small molecules blocking the p53/MDM2 interaction. The Mdm2 inhibitor Nutlin-3 is restricted to tumors carrying wtp53. In contrast, RITA, a compound that binds p53, has recently been shown also to restore transcriptional functions of mtp53. As more than 50% of solid tumors carry p53 mutations, RITA promises to be a more effective therapeutic strategy than Nutlin-3. We investigated effects of RITA on apoptosis, cell cycle and induction of 45 p53 target genes in a panel of 14 cell lines from different tumor entities with different p53 status as well as primary lymphocytes and fibroblasts. Nine cell strains expressed wtp53, four harbored mtp53, and three were characterized by the loss of p53 protein. A significant induction of cell death upon RITA was observed in 7 of 16 cell lines. The nonmalignant cells in our panel were substantially less sensitive. We found that in contrast to Nultin-3, RITA is capable to induce cell death not only in tumor cells harboring wtp53 and mtp53 but also in p53-null cells. Importantly, whereas p53 has a central role for RITA-mediated effects in wtp53 cells, neither p53 nor p63 or p73 were essential for the RITA response in mtp53 or p53-null cells in our panel demonstrating that besides the known p53-dependent action of RITA in wtp53 cells, RITA can induce cell death also independently of p53 in cells harboring defective p53. We identified an important role of both p38 and JNK/SAPK for sensitivity to RITA in these cells leading to a typical caspase- and BAX/BAK-dependent mitochondrial apoptosis. In conclusion, our data demonstrate that RITA can induce apoptosis through p38 and JNK/SAPK not only in tumor cells harboring wtp53 and mtp53 but also in p53-null cells, making RITA an interesting tumor-selective drug. | Western Blotting | | 25010984
|
The detergent-soluble cytoplasmic pool of survivin suppresses anoikis and its expression is associated with metastatic disease of human colon cancer.
Hori, M; Miki, T; Okamoto, M; Yazama, F; Konishi, H; Kaneko, H; Shimamoto, F; Ota, T; Temme, A; Tatsuka, M
PloS one
8
e55710
2013
Mostrar resumen
Survivin is a component of the chromosomal passenger complex (CPC) that is essential for accurate chromosome segregation. Interfering with the function of Survivin in mitosis leads to chromosome segregation errors and defective cytokinesis. Survivin contains a Baculovirus IAP Repeat (BIR) and therefore was originally classified as inhibitor of apopotosis protein (IAP), yet its role in apoptosis after cellular stress remains largely unknown. We demonstrate here, that Survivin predominantly suppresses anoikis, a form of programmed cell death induced by loss of cellular adhesion to extracellular matrix. Interestingly, cells ectopically overexpressing EGFP-Survivin showed after loss of cell-matrix-interaction a decreased expression of IκB-α. Subsequent subcellular protein fractionation and immunoprecipitation experiments revealed that XIAP interacts with detergent-soluble Survivin which is known to cooperatively activate NF-κB signaling. Examination of the expression levels of detergent soluble Survivin in colorectal cancer cell lines and in colorectal cancerous tissues revealed that detergent soluble cytoplasmic Survivin levels correlated inversely with anoikis susceptibility in colorectal cancer. Therefore, the detergent soluble cytoplasmic Survivin might be a promising predictive biomarker for lymph node and distant metastases of colorectal cancer. We conclude that an anti-apoptotic function of detergent-soluble Survivin in interphase cells experiencing anoikis is mediated at least via XIAP/IκB-α/NF-κB signaling. | Western Blotting | | 23405201
|
Pregnenolone sulfate activates basic region leucine zipper transcription factors in insulinoma cells: role of voltage- gated Ca2+ channels and transient receptor potential melastatin 3 channels.
Müller I, Rössler OG, Thiel G
Molecular pharmacology
80
1179-89. Epub 2011 Sep 23.
2010
Mostrar resumen
The neurosteroid pregnenolone sulfate activates a signaling cascade in insulinoma cells involving activation of extracellular signal-regulated protein kinase and enhanced expression of the transcription factor Egr-1. Here, we show that pregnenolone sulfate stimulation leads to a significant elevation of activator protein-1 (AP-1) activity in insulinoma cells. Expression of the basic region leucine zipper (bZIP) transcription factors c-Jun and c-Fos is up-regulated in insulinoma cells and pancreatic β-cells in primary culture after pregnenolone sulfate stimulation. Up-regulation of a chromatin-embedded c-Jun promoter/luciferase reporter gene transcription in pregnenolone sulfate-stimulated insulinoma cells was impaired when the AP-1 binding sites were mutated, indicating that these motifs function as pregnenolone sulfate response elements. In addition, phosphorylation of cAMP response element (CRE)-binding protein is induced and transcription of a CRE-controlled reporter gene is stimulated after pregnenolone sulfate treatment, indicating that the CRE functions as a pregnenolone sulfate response element as well. Pharmacological and genetic experiments revealed that both L-type Ca(2+) channels and transient receptor potential melastatin 3 (TRPM3) channels are essential for connecting pregnenolone sulfate stimulation with enhanced AP-1 activity and bZIP-mediated transcription in insulinoma cells. In contrast, pregnenolone sulfate stimulation did not enhance AP-1 activity or c-Jun and c-Fos expression in pituitary corticotrophs that express functional L-type Ca(2+) channels but only trace amounts of TRPM3. We conclude that expression of L-type Ca(2+) channels is not sufficient to activate bZIP-mediated gene transcription by pregnenolone sulfate. Rather, additional expression of TRPM3 or depolarization of the cells is required to connect pregnenolone sulfate stimulation with enhanced gene transcription. | | | 21948387
|
Immediate-early transcriptional response to angiotensin II in human adrenocortical cells.
Thiel, G; Rössler, OG
Endocrinology
152
4211-23
2010
Mostrar resumen
Angiotensin II binds to the angiotensin II receptors type 1 (AT1 receptors) in adrenocortical cells and triggers an intracellular signaling cascade leading to changes in the gene expression pattern. Here, we show that stimulation with angiotensin II induces the expression of biologically active early growth response (Egr)-1, a zinc finger transcription factor, in human H295R adrenocortical cells. Expression of a dominant-negative mutant of the ternary complex factor Elk-1, a key transcriptional regulator of serum response element-driven gene transcription, prevented Egr-1 expression in angiotensin II-stimulated H295R cells, indicating that Ets-like protein-1 (Elk-1) or related ternary complex factors connect the intracellular signaling cascade elicited by activation of AT1 receptors with transcription of the Egr-1 gene. These data were corroborated by the fact that angiotensin II stimulation increased the transcription activation potential of Elk-1. In addition, activator protein-1 transcriptional activity was significantly elevated in angiotensin II-treated H295R cells. Expression of c-Jun and c-Fos was increased as well as the transcription activation potential of c-Fos. Expression of a dominant-negative mutant of Elk-1 reduced c-Fos expression in angiotensin II-stimulated adrenocortical cells, suggesting that the serum response element within the c-Fos promoter functions as an angiotensin II-response element. Expression of a dominant-negative mutant of c-Jun reduced activator protein-1 activity in angiotensin II-stimulated adrenocortical cells and reduced the up-regulation of c-Jun after angiotensin II stimulation. Thus, c-Jun regulates its own expression in adrenocortical cells. Together, the data show that angiotensin II stimulation activates the transcription factors Egr-1, Elk-1, c-Jun, and c-Fos in adrenocortical cells, leading to stimulus-dependent changes in the gene expression pattern. | | | 21914770
|
Thapsigargin induces expression of ATF3 in human keratinocytes involving Ca2+ ions and c-Jun N-terminal protein kinase.
Spohn D, Rossler OG, Philipp SE, Raubuch M, Kitajima S, Griesemer D, Hoth M, Thiel G
Mol Pharmacol
2009
Mostrar resumen
Thapsigargin is a specific inhibitor of the SERCA ATPase of the endoplasmic reticulum. Here, we show that stimulation of human HaCaT keratinocytes with nanomolar concentrations of thapsigargin triggers expression of ATF3, a basic-region leucin zipper transcription factor. ATF3 expression was also upregulated in thapsigargin-stimulated glioma cells, hepatoma cells, retinal pigment epithelial cells, and airway epithelial cells. Thapsigargin-induced upregulation of ATF3 expression in keratinocytes was attenuated by BAPTA-AM, or by expression of the Ca(2+)-binding protein parvalbumin in the cytosol of HaCaT cells, but not by a panel of pharmacological agents that chelate extracellular Ca(2+) (EGTA) or inhibit either ryanodine receptors (dantrolene) or voltage-gated Ca(2+) channels (nifedipine). Hence, elevated levels of intracellular Ca(2+), released from intracellular stores, are essential for the effect of thapsigargin on the biosynthesis of ATF3. The thapsigargin-induced signaling pathway was blocked by expression of either MAP kinase phophatases-1 or -5. Experiments involving pharmacological and genetic tools revealed the importance of c-Jun N-terminal protein kinase (JNK) within the signaling cascade, while inhibition of extracellular signal-regulated protein kinase or p38 protein kinase did not attenuate thapsigargin-induced expression of ATF3. Functional studies showed that treatment of HaCaT keratinocytes with thapsigargin led to a 2-fold induction of caspase-3/7 activity. The upregulation of caspase-3/7 activity in thapsigargin-stimulated HaCaT cells was attenuated by inhibition of JNK. Together, these data show that stimulation of HaCaT cells with thapsigargin induces a specific signaling pathway in keratinocytes involving activation of JNK, biosynthesis of ATF3, and upregulation of caspase-3/7 activity. | | | 20713550
|
c-Jun N-terminal kinase binding domain-dependent phosphorylation of mitogen-activated protein kinase kinase 4 and mitogen-activated protein kinase kinase 7 and balancing cross-talk between c-Jun N-terminal kinase and extracellular signal-regulated kinase pathways in cortical neurons.
M Repici,L Mare,A Colombo,C Ploia,A Sclip,C Bonny,P Nicod,M Salmona,T Borsello
Neuroscience
159
2009
Mostrar resumen
The c-Jun N-terminal kinase (JNK) is a mitogen-activated protein kinase (MAPK) activated by stress-signals and involved in many different diseases. Previous results proved the powerful effect of the cell permeable peptide inhibitor d-JNKI1 (d-retro-inverso form of c-Jun N-terminal kinase-inhibitor) against neuronal death in CNS diseases, but the precise features of this neuroprotection remain unclear. We here performed cell-free and in vitro experiments for a deeper characterization of d-JNKI1 features in physiological conditions. This peptide works by preventing JNK interaction with its c-Jun N-terminal kinase-binding domain (JBD) dependent targets. We here focused on the two JNK upstream MAPKKs, mitogen-activated protein kinase kinase 4 (MKK4) and mitogen-activated protein kinase kinase 7 (MKK7), because they contain a JBD homology domain. We proved that d-JNKI1 prevents MKK4 and MKK7 activity in cell-free and in vitro experiments: these MAPKK could be considered not only activators but also substrates of JNK. This means that d-JNKI1 can interrupt downstream but also upstream events along the JNK cascade, highlighting a new remarkable feature of this peptide. We also showed the lack of any direct effect of the peptide on p38, MEK1, and extracellular signal-regulated kinase (ERK) in cell free, while in rat primary cortical neurons JNK inhibition activates the MEK1-ERK-Ets1/c-Fos cascade. JNK inhibition induces a compensatory effect and leads to ERK activation via MEK1, resulting in an activation of the survival pathway-(MEK1/ERK) as a consequence of the death pathway-(JNK) inhibition. This study should hold as an important step to clarify the strong neuroprotective effect of d-JNKI1. | | | 19135136
|
Inhibition of apoptosome activation protects injured motor neurons from cell death.
Kanungo, AK; Hao, Z; Elia, AJ; Mak, TW; Henderson, JT
The Journal of biological chemistry
283
22105-12
2008
Mostrar resumen
Within the mammalian central nervous system many forms of neurodegenerative injury are regulated via programmed cell death, a highly conserved program of cellular suicide. Programmed cell death is regulated by multiple signaling pathways, which have been identified within mammalian cells, although several lines of evidence suggest that the intrinsic pathway predominantly regulates the death of motor neurons following acute injury in vivo. We have tested this hypothesis by performing facial axotomies on cytochrome c knock-in mice containing a point mutation in the genomic locus of cytochrome c resulting in a lysine to alanine conversion at position 72 of the protein. The introduced mutation inhibits the ability of cytochrome c to induce the formation of the apoptosome, a protein complex that is principally required for the activation of the intrinsic pathway, but does not alter its function in oxidative phosphorylation. Homozygous cytochrome c knock-in mutants displayed a significant enhancement in motor neuron survival following injury when compared with littermate controls, thus establishing the apoptosome as a viable target for protecting motor neurons from neural injury. However, protection of facial motor neurons differs from that previously reported in mice either overexpressing anti-apoptotic or lacking pro-apoptotic members of the Bcl-2 family, which are thought to regulate several aspects of mitochondrial dysfunction including the release of cytochrome c from the mitochondria to the cytoplasm. Therefore, these results directly demonstrate for the first time the influence of the apoptosome on injury-induced neuronal programmed cell death in vivo isolated from upstream Bcl-2 family-mediated effects. | Immunohistochemistry | | 18550520
|
Transforming signals resulting from sustained activation of the PDGFbeta receptor in mortal human fibroblasts.
Petti, Lisa M, et al.
J. Cell. Sci., 121: 1172-82 (2008)
2008
Mostrar resumen
The platelet-derived growth factor beta receptor (PDGFbetaR) plays an important role in proliferation and motility of fibroblasts. We have been investigating the effects of sustained PDGFbetaR activation in mortal human diploid fibroblasts (HDFs), which are typically difficult to transform. We have previously shown that the bovine papillomavirus E5 protein, through its ability to crosslink and constitutively activate the PDGFbetaR, induces morphological transformation, enhanced growth and loss of contact inhibition (focus formation) in HDFs. Here, we characterized two E5 mutants as being severely defective for focus formation but still competent for enhanced growth, suggesting that proliferation is insufficient for loss of contact inhibition. These E5 mutants were then used in a comparative study to distinguish the PDGFbetaR signaling intermediates required for the enhanced growth phenotype from those required for focus formation. Our data suggested that a PI 3-kinase (PI3K)-AKT-cyclin D3 pathway, a Grb2-Gab1-SHP2 complex and JNK played a role in the enhanced growth phenotype. However, a SHP2-p66Shc-p190BRhoGAP complex and ROCK were implicated exclusively in focus formation. We speculate that a SHP2-p66Shc-p190BRhoGAP signaling complex recruited to the activated PDGFbetaR promotes a distinct Rho-dependent process required for focus formation but not growth of HDFs. | | Human | 18349076
|
Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin action in skeletal muscle cells.
Weigert, C; Kron, M; Kalbacher, H; Pohl, AK; Runge, H; Häring, HU; Schleicher, E; Lehmann, R
Molecular endocrinology (Baltimore, Md.)
22
2729-40
2008
Mostrar resumen
Transduction of the insulin signal is mediated by multisite Tyr and Ser/Thr phosphorylation of the insulin receptor substrates (IRSs). Previous studies on the function of single-site phosphorylation, particularly phosphorylation of Ser-302, -307, and -318 of IRS-1, showed attenuating as well as enhancing effects on insulin action. In this study we investigated a possible cross talk of these opposedly acting serine residues in insulin-stimulated skeletal muscle cells by monitoring phosphorylation kinetics, and applying loss of function, gain of function, and combination mutants of IRS-1. The phosphorylation at Ser-302 was rapid and transient, followed first by Ser-318 phosphorylation and later by phosphorylation of Ser-307, which remained elevated for 120 min. Mutation of Ser-302 to alanine clearly reduced the subsequent protein kinase C-zeta-mediated Ser-318 phosphorylation. The Ser-307 phosphorylation was independent of Ser-302 and/or Ser-318 phosphorylation status. The functional consequences of these phosphorylation patterns were studied by the expression of IRS-1 mutants. The E302A307E318 mutant simulating the early phosphorylation pattern resulted in a significant increase in Akt and glycogen synthase kinase 3 phosphorylation. Furthermore, glucose uptake was enhanced. Because the down-regulation of the insulin signal was not affected, this phosphorylation pattern seems to be involved in the enhancement but not in the termination of the insulin signal. This enhancing effect was completely absent when Ser-302 was unphosphorylated and Ser-307 was phosphorylated as simulated by the A302E307E318 mutant. Phospho-Ser-318, sequentially phosphorylated at least by protein kinase C-zeta and a mammalian target of rapamycin/raptor-dependent kinase, was part of the positive as well as of the subsequent negative phosphorylation pattern. Thus we conclude that insulin stimulation temporally generates different phosphorylation statuses of the same residues that exert different functions in insulin signaling. | | | 18927238
|