



ALP10-KC Sigma-AldrichApolipoprotein A-I, human (1mg) KC


Prodotti consigliati






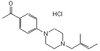

Panoramica
| Replacement Information |
|---|
| References |
|---|
| Product Information | |
|---|---|
| Presentation | Liquid in 10 mM NH4HCO3, pH 7.4. Non-sterile, no preservatives. |
| Quality Level | MQ300 |
| Applications | |
|---|---|
| Key Applications |
|
| Physicochemical Information |
|---|
| Dimensions |
|---|
| Materials Information |
|---|
| Toxicological Information |
|---|
| Safety Information according to GHS |
|---|
| Safety Information |
|---|
| Packaging Information | |
|---|---|
| Material Size | Bulk packaging. Please inquire. |
| Transport Information |
|---|
| Supplemental Information |
|---|
| Specifications |
|---|
| Global Trade Item Number | |
|---|---|
| Numero di catalogo | GTIN |
| ALP10-KC | 04053252482571 |