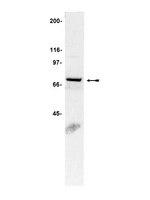
Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells.
Chong, LW; Hsu, YC; Lee, TF; Lin, Y; Chiu, YT; Yang, KC; Wu, JC; Huang, YT
BMC gastroenterology
15
22
2015
Mostra il sommario
Non-alcoholic steatohepatitis (NASH) is associated with hepatic fibrogenesis. Despite well-known cholesterol-lowering action of statins, their mechanisms against NASH-mediated fibrogenesis remain unclear. This study aimed at investigating the in vitro and in vivo anti-fibrotic properties of fluvastatin (Flu).Palmitate (PA)-induced changes in intracellular hydrogen peroxide levels in primary rat hepatocytes (PRHs) and human hepatoma cell line (HepG2) were quantified by dichlorofluorescein diacetate (DCF-DA) dye assay, whereas changes in expressions of NADPH oxidase gp91 (phox) subunit, α-smooth muscle actin (α-SMA), and NFκB p65 nuclear translocation were quantified with Western blotting. Quantitative real-time polymerase chain reaction (q-PCR) was used to investigate mRNA expressions of pro-inflammatory genes (ICAM-1, IL-6, TNF-α). Conditioned medium (CM) from PA-treated PRHs was applied to cultured rat hepatic stellate cell line, HSC-T6, with or without Flu-pretreatment for 2 h. Pro-fibrogenic gene expressions (COL1, TIMP-1, TGF-β1, α-SMA) and protein expression of α-SMA were analyzed. In vivo study using choline-deficient L-amino acid defined (CDAA) diet-induced rat NASH model was performed by randomly assigning Wistar rats (n = 28) to normal controls (n = 4), CDAA diet with vehicles, and CDAA diet with Flu (5 mg/kg or 10 mg/kg) (n = 8 each) through gavage for 4 or 8 weeks. Livers were harvested for histological, Western blot (α-SMA), and q-PCR analyses for expressions of pro-inflammatory (IL-6, iNOS, ICAM-1) and pro-fibrogenic (Col1, α-SMA, TIMP-1) genes.In vitro, Flu (1-20 μM) inhibited PA-induced free-radical production, gp91 (phox) expression, and NFκB p65 translocation in HepG2 and PRHs, while CM-induced α-SMA protein expression and pro-fibrogenic gene expressions in HSC-T6 were suppressed in Flu-pretreated cells compared to those without pretreatment. Moreover, α-SMA protein expression was significantly decreased in HSC-T6 cultured with CM from PA-Flu-treated PRHs compared to those cultured with CM from PA-treated PRHs. Flu also reduced steatosis and fibrosis scores, α-SMA protein expression, mRNA expression of pro-inflammatory and pro-fibrogenic genes in livers of CDAA rats.We demonstrated PA-induced HSC activation through paracrine effect of hepatocyte in vitro that was significantly suppressed by pre-treating HSC with Flu. In vivo, Flu alleviated steatosis-induced HSC activation and hepatic fibrogenesis through mitigating inflammation and oxidative stress, suggesting possible therapeutic role of Flu against NASH. | | 25886887
 |
Acute ethanol induces apoptosis by stimulating TRPC6 via elevation of superoxide in oxygenated podocytes.
Lu, XY; Liu, BC; Wang, LH; Yang, LL; Bao, Q; Zhai, YJ; Alli, AA; Thai, TL; Eaton, DC; Wang, WZ; Ma, HP
Biochimica et biophysica acta
1853
965-74
2015
Mostra il sommario
Our recent studies indicate that hydrogen peroxide (H2O2) only at high concentrations can cause oxidative stress in renal epithelial cells and induce apoptosis of podocytes. Consistently, the present study shows that H2O2, even at 1 mM, failed to induce intracellular oxidative stress and apoptosis of the podocytes due to efficient activity of catalase, an enzyme which degrades H2O2 to produce water and oxygen (O2). However, H2O2 acted as a source of O2 to allow acute ethanol to induce superoxide production and cause apoptosis of the podocytes. In contrast, acute ethanol alone did not elevate intracellular superoxide, even though it stimulates expression and translocation of p47phox to the plasma membrane. Inhibition of catalase abolished not only O2 production from H2O2 degradation, but also NOX2-dependent superoxide production in the podocytes challenged by both H2O2 and acute ethanol. In parallel, acute ethanol in the presence of H2O2, but neither ethanol nor H2O2 alone, stimulated transient receptor potential canonical 6 (TRPC6) channels and caused TRPC6-dependent elevation of intracellular Ca2+. These data suggest that exogenous H2O2 does not induce oxidative stress due to rapid degradation to produce O2 in the podocytes, but the oxygenated podocytes become sensitive to acute ethanol challenge and undergo apoptosis via a TRPC6-dependent elevation of intracellular Ca2+. Since cultured podocytes are considered in hypoxic conditions, H2O2 may be used as a source of O2 to establish an ischemia-reperfusion model in some type of cultured cells in which H2O2 does not directly induce intracellular oxidative stress. | | 25601712
|
Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression.
Song, X; Liu, BC; Lu, XY; Yang, LL; Zhai, YJ; Eaton, AF; Thai, TL; Eaton, DC; Ma, HP; Shen, BZ
Biochimica et biophysica acta
1843
894-901
2014
Mostra il sommario
Clinical evidence suggests that statins reduce cancer incidence and mortality. However, there is lack of in vitro data to show the mechanism by which statins can reduce the malignancies of cancer cells. We used a human B lymphoma Daudi cells as a model and found that lovastatin inhibited, whereas exogenous cholesterol (Cho) stimulated, proliferation cell cycle progression in control Daudi cells, but not in the cells when transient receptor potential canonical 6 (TRPC6) channel was knocked down. Lovastatin decreased, whereas Cho increased, the levels of intracellular reactive oxygen species (ROS) respectively by decreasing or increasing the expression of p47-phox and gp91-phox (NOX2). Reducing intracellular ROS with either a mimetic superoxide dismutase (TEMPOL) or an NADPH oxidase inhibitor (apocynin) inhibited cell proliferation, particularly in Cho-treated cells. The effects of TEMPOL or apocynin were mimicked by inhibition of TRPC6 with SKF-96365. Lovastatin decreased TRPC6 expression and activity via a Cho-dependent mechanism, whereas Cho increased TRPC6 expression and activity via an ROS-dependent mechanism. Consistent with the fact that TRPC6 is a Ca(2+)-permeable channel, lovastatin decreased, but Cho increased, intracellular Ca(2+) also via ROS. These data suggest that lovastatin inhibits malignant B cell proliferation by reducing membrane Cho, intracellular ROS, TRPC6 expression and activity, and intracellular Ca(2+). | | 24518247
|
Angiotensin Receptor Mediated Oxidative Stress Is Associated with Impaired Cardiac Redox Signaling and Mitochondrial Function in Insulin Resistant Rats.
Vazquez-Medina, Jose Pablo, et al.
Am. J. Physiol. Heart Circ. Physiol., (2013)
2013
Mostra il sommario
Activation of angiotensin receptor type 1 (AT1) contributes to NADPH oxidase (Nox)-derived oxidative stress during metabolic syndrome. However, AT1 activation in modulating redox signaling, mitochondrial function and oxidative stress in the heart during metabolic syndrome remains more elusive. To test the hypothesis that AT1 activation increases oxidative stress while impairing redox signaling and mitochondrial function in the heart during insulin resistant conditions, diet-induced obese insulin resistant (OLETF) rats (n=8/group) were treated with the AT1 blocker (ARB) Olmesartan for 6 weeks. Cardiac Nox2 protein expression increased 40% in OLETF compared to age-matched LETO rats while mRNA and protein expression of the H2O2-producing, Nox4, increased 40-100%. ARB treatment prevented the increase in Nox2 without altering Nox4. ARB treatment also normalized increased levels of protein and lipid oxidation (nitrotyrosine, 4-hydroxynonenal) and increased the redox-sensitive transcription factor, Nrf2, by 30%, and antioxidant enzymes (SOD, catalase, GPx) by 50-70%. Citrate synthase (CS) and succinate dehydrogenase (SDH) activities decreased 60-70% while cardiac succinate levels decreased 35% in OLETF compared to LETO suggesting that mitochondrial function in the heart is impaired during insulin resistance. ARB treatment normalized CS, SDH and succinate levels while increasing AMPK phosphorylation and normalizing AKT phosphorylation suggesting that AT1 activation impairs cellular metabolism in the diabetic heart. These data suggest that the cardiovascular complications associated with metabolic syndrome may be the consequence of oxidants produced as a consequence of AT1-mediated Nox2 activation that is associated with impaired redox signaling and mitochondrial activity, resulting in dysregulation of cellular metabolism in the heart. - | | 23771688
|
Advanced oxidation protein products induce cardiomyocyte death via Nox2/Rac1/superoxide-dependent TRAF3IP2/JNK signaling.
Valente, AJ; Yoshida, T; Clark, RA; Delafontaine, P; Siebenlist, U; Chandrasekar, B
Free radical biology & medicine
60
125-35
2013
Mostra il sommario
Advanced oxidation protein products (AOPPs) are formed during chronic oxidative stress as a result of reactions between plasma proteins and chlorinated oxidants. Their levels are elevated during various cardiovascular diseases. Because elevated AOPPs serve as independent risk factors for ischemic heart disease, and cardiomyocyte death is a hallmark of ischemic heart disease, we hypothesized that AOPPs will induce cardiomyocyte death. AOPP-modified mouse serum albumin (AOPP-MSA) induced significant death of neonatal mouse cardiomyocytes that was attenuated by knockdown of the receptor for advanced glycation end products, but not CD36. Notably, TRAF3-interacting protein 2 (TRAF3IP2; also known as CIKS or Act1) knockdown blunted AOPP-induced apoptosis. AOPP-MSA stimulated Nox2/Rac1-dependent superoxide generation, TRAF3IP2 expression, and TRAF3IP2-dependent JNK activation. The superoxide anion generating xanthine/xanthine oxidase system and hydrogen peroxide both induced TRAF3IP2 expression. Further, AOPP-MSA induced mitochondrial Bax translocation and release of cytochrome c into cytoplasm. Moreover, AOPP-MSA suppressed antiapoptotic Bcl-2 and Bcl-xL expression. These effects were reversed by TRAF3IP2 knockdown or forced expression of mutant JNK. Similar to its effects in neonatal cardiomyocytes, AOPP-MSA induced adult cardiomyocyte death in part via TRAF3IP2. These results demonstrate for the first time that AOPPs induce cardiomyocyte death via Nox2/Rac1/superoxide-dependent TRAF3IP2/JNK activation in vitro and suggest that AOPPs may contribute to myocardial injury in vivo. Thus TRAF3IP2 may represent a potential therapeutic target in ischemic heart disease. | | 23453926
|
Stress reaction in outer segments of photoreceptors after blue light irradiation.
Roehlecke, C; Schumann, U; Ader, M; Brunssen, C; Bramke, S; Morawietz, H; Funk, RH
PloS one
8
e71570
2013
Mostra il sommario
The retina is prone to oxidative stress from many factors which are also involved in the pathogenesis of degenerative diseases. In this study, we used the application of blue light as a physiological stress factor. The aim of this study was to identify the major source of intracellular ROS that mediates blue light-induced detrimental effects on cells which may lead to cytotoxicity. We hypothesized that outer segments are the major source of blue light induced ROS generation. In photoreceptors, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) enzymes and the recently found respiratory chain complexes may represent a major source for reactive oxygen species (ROS), beside mitochondria and chromophores. Therefore, we investigated this hypothesis and analysed the exact localization of the ROS source in photoreceptors in an organotypic culture system for mouse retinas. Whole eyeball cultures were irradiated with visible blue light (405 nm) with an output power of 1 mW/cm². Blue light impingement lead to an increase of ROS production (detected by H2DCFDA in live retinal explants), which was particularly strong in the photoreceptor outer segments. Nox-2 and Nox-4 proteins are sources of ROS in blue light irradiated photoreceptors; the Nox inhibitor apocynin decreased ROS stimulated by blue light. Concomitantly, enzyme SOD-1, a member of the antioxidant defense system, indicator molecules of protein oxidation (CML) and lipid oxidation (MDA and 4-HNE) were also increased in the outer segments. Interestingly, outer segments showed a mitochondrial-like membrane potential which was demonstrated using two dyes (JC-1 and TMRE) normally exclusively associated with mitochondria. As in mitochondria, these dyes indicated a decrease of the membrane potential in hypoxic states or cell stress situations. The present study demonstrates that ROS generation and oxidative stress occurs directly in the outer segments of photoreceptors after blue light irradiation. | Western Blotting | 24039718
|
H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling.
Woolley, JF; Naughton, R; Stanicka, J; Gough, DR; Bhatt, L; Dickinson, BC; Chang, CJ; Cotter, TG
PloS one
7
e34050
2011
Mostra il sommario
The internal tandem duplication (ITD) of the juxtamembrane region of the FLT3 receptor has been associated with increased reactive oxygen species (ROS) generation in acute myeloid leukemia (AML). How this elevated level of ROS contributes to the leukemic phenotype, however, remains poorly understood. In this work we show that ROS in the FLT3-ITD expressing AML cell line MV4-11 is reduced by treatment with PKC412, an inhibitor of FLT3, DPI, a flavoprotein inhibitor, and VAS2870, a Nox specific inhibitor, suggesting that ROS production is both FLT3 and NADPH oxidase dependent. The majority of these ROS co-localize to the endoplasmic reticulum (ER), as determined with the H(2)O(2)-specific aryl-boronate dye Peroxyorange 1, which also corresponds to co-localization of p22phox. Moreover, knocking down p22phox dramatically reduces H(2)O(2) after 24 hours in the ER, without affecting mitochondrial ROS. Significantly, the FLT3 inhibitor PKC412 reduces H(2)O(2) in FLT3-ITD expressing cell lines (MV4-11, MOLM-13) through reduction of p22phox over 24 hours. Reduced p22phox is achieved by proteasomal degradation and is prevented upon GSK3-β inhibition. Knockdown of p22phox resulted in reduced STAT5 signalling and reduced Pim-1 levels in the cells after 24 hours. Thus, we have shown that FLT3 driven H(2)O(2) production in AML cells is mediated by p22phox and is critical for STAT5 signalling. | | 22807997
|
Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy.
Shanmugam, P; Valente, AJ; Prabhu, SD; Venkatesan, B; Yoshida, T; Delafontaine, P; Chandrasekar, B
Journal of molecular and cellular cardiology
50
928-38
2010
Mostra il sommario
Angiotensin-II (Ang-II) plays a key role in myocardial hypertrophy, remodeling and failure. We investigated whether Ang-II-induced cardiomyocyte hypertrophy is dependent on WNT1 inducible signaling pathway protein 1 (WISP1), a pro growth factor. Ang-II induced hypertrophy and WISP1 expression in neonatal rat cardiomyocytes (NRCM), effects that were significantly inhibited by pre-treatment with the AT1 antagonist losartan and by WISP1 knockdown. Further, Ang-II induced WISP1 was superoxide-dependent, and inhibited by DPI, an inhibitor of NADPH oxidases, and by knockdown of NOX2. AT1 was physically associated with NOX2 both in vitro and in vivo, and Ang-II increased this interaction in vivo. Ang-II induced WISP1 expression via superoxide/Akt/GSK3β/β-catenin/TCF/LEF and by Akt-dependent CREB activation. Further, Ang-II also activated CREB via superoxide-mediated p38 MAPK and ERK activation. Continuous infusion of Ang-II for 7days induced myocardial hypertrophy in rats, and was associated with increased Akt, p-Akt, p-p38 MAPK, p-ERK1/2, and WISP1 expression. These results demonstrate that Ang-II induced cardiomyocyte hypertrophy is mediated through AT1, NOX2 and the induction of WISP1, and may involve the direct interaction of AT1 with NOX2. Thus targeting both WISP1 and NOX2 may have a therapeutic potential in improving cardiomyocyte survival and growth following myocardial injury and remodeling. This article is part of a Special Issue entitled 'Possible Editorial'. | Western Blotting | 21376054
|
NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia.
Yoshioka, H; Niizuma, K; Katsu, M; Okami, N; Sakata, H; Kim, GS; Narasimhan, P; Chan, PH
Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism
31
868-80
2010
Mostra il sommario
Medium spiny neurons (MSNs) constitute most of the striatal neurons and are known to be vulnerable to ischemia; however, the mechanisms of the vulnerability remain unclear. Activated forms of nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase (NOX), which require interaction between cytosolic and membrane-bound subunits, are among the major sources of superoxide in the central nervous system. Although increasing evidence suggests that NOX has important roles in neurodegenerative diseases, its roles in MSN injury after transient global cerebral ischemia (tGCI) have not been elucidated. To clarify this issue, C57BL/6 mice were subjected to tGCI by bilateral common carotid artery occlusion for 22 minutes. Western blot analysis revealed upregulation of NOX subunits and recruitment of cytosolic subunits to the cell membrane at early (3 to 6 hours) and late (72 hours) phases after tGCI. Taken together with immunofluorescent studies, this activation arose in MSNs and endothelial cells at the early phase, and in reactive microglia at the late phase. Pharmacological and genetic inhibition of NOX attenuated oxidative injury, microglial activation, and MSN death after tGCI. These findings suggest that NOX has pivotal roles in MSN injury after tGCI and could be a therapeutic target for brain ischemia. Testo completo dell'articolo | | 20859296
|
Redox survival signalling in retina-derived 661W cells.
Mackey, AM; Sanvicens, N; Groeger, G; Doonan, F; Wallace, D; Cotter, TG
Cell death and differentiation
15
1291-303
2008
Mostra il sommario
Reactive oxygen species have been implicated in processes involving cellular damage and subsequent cell death, especially in organs such as the eye that are constantly exposed to excitatory signals. However, recent studies have shown that oxidant species can also act as intracellular signalling molecules promoting cell survival, but little is known about this mechanism in the retina. The present study demonstrates for the first time that hydrogen peroxide (H2O2) is generated rapidly and acts as a pro-survival signal in response to a variety of apoptotic stimuli in retina-derived 661W cells and in the retinal ganglion cell line RGC-5. Focussing on 661Ws and serum deprivation, we systematically investigated pro-survival and pro-death pathways and discovered that the rapid and transient burst of H2O2 activates the AKT survival pathway. Activation of the apoptotic machinery takes place following the decline of H2O2 to basal levels. To substantiate this proposed pro-survival role of H2O2, we inhibited the oxidant burst, which exacerbated cell death. Conversely, maintenance of the oxidant signal using exogenous H2O2 enhanced cell survival. Overall, the results presented in this study provide evidence for a novel role of H2O2 in mediating survival of retinal cells in response to apoptotic stimuli. | | 18404155
|