Coupled local translation and degradation regulate growth cone collapse.
Deglincerti, A; Liu, Y; Colak, D; Hengst, U; Xu, G; Jaffrey, SR
Nature communications
6
6888
2015
Mostra il sommario
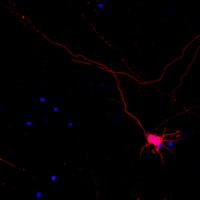
Local translation mediates axonal responses to Semaphorin3A (Sema3A) and other guidance cues. However, only a subset of the axonal proteome is locally synthesized, whereas most proteins are trafficked from the soma. The reason why only specific proteins are locally synthesized is unknown. Here we show that local protein synthesis and degradation are linked events in growth cones. We find that growth cones exhibit high levels of ubiquitination and that local signalling pathways trigger the ubiquitination and degradation of RhoA, a mediator of Sema3A-induced growth cone collapse. Inhibition of RhoA degradation is sufficient to remove the protein-synthesis requirement for Sema3A-induced growth cone collapse. In addition to RhoA, we find that locally translated proteins are the main targets of the ubiquitin-proteasome system in growth cones. Thus, local protein degradation is a major feature of growth cones and creates a requirement for local translation to replenish proteins needed to maintain growth cone responses. | | | 25901863
 |
Regulation of axon growth by the JIP1-AKT axis.
Dajas-Bailador, F; Bantounas, I; Jones, EV; Whitmarsh, AJ
Journal of cell science
127
230-9
2014
Mostra il sommario
The polarisation of developing neurons to form axons and dendrites is required for the establishment of neuronal connections leading to proper brain function. The protein kinase AKT and the MAP kinase scaffold protein JNK-interacting protein-1 (JIP1) are important regulators of axon formation. Here we report that JIP1 and AKT colocalise in axonal growth cones of cortical neurons and collaborate to promote axon growth. The loss of AKT protein from the growth cone results in the degradation of JIP1 by the proteasome, and the loss of JIP1 promotes a similar fate for AKT. Reduced protein levels of both JIP1 and AKT in the growth cone can be induced by glutamate and this coincides with reduced axon growth, which can be rescued by a stabilized mutant of JIP1 that rescues AKT protein levels. Taken together, our data reveal a collaborative relationship between JIP1 and AKT that is required for axon growth and can be regulated by changes in neuronal activity. | | | 24198394
|
Gαz regulates BDNF-induction of axon growth in cortical neurons.
Hultman, R; Kumari, U; Michel, N; Casey, PJ
Molecular and cellular neurosciences
58
53-61
2014
Mostra il sommario
The disruption of neurotransmitter and neurotrophic factor signaling in the central nervous system (CNS) is implicated as the root cause of neuropsychiatric disorders, including schizophrenia, epilepsy, chronic pain, and depression. Therefore, identifying the underlying molecular mechanisms by which neurotransmitter and neurotrophic factor signaling regulates neuronal survival or growth may facilitate identification of more effective therapies for these disorders. Previously, our lab found that the heterotrimeric G protein, Gz, mediates crosstalk between G protein-coupled receptors and neurotrophin signaling in the neural cell line PC12. These data, combined with Gαz expression profiles--predominantly in neuronal cells with higher expression levels corresponding to developmental times of target tissue innervation--suggested that Gαz may play an important role in neurotrophin signaling and neuronal development. Here, we provide evidence in cortical neurons, both manipulated ex vivo and those cultured from Gz knockout mice, that Gαz is localized to axonal growth cones and plays a significant role in the development of axons of cortical neurons in the CNS. Our findings indicate that Gαz inhibits BDNF-stimulated axon growth in cortical neurons, establishing an endogenous role for Gαz in regulating neurotrophin signaling in the CNS. | | | 24321455
|
Specificity of anti-tau antibodies when analyzing mice models of Alzheimer's disease: problems and solutions.
Petry, FR; Pelletier, J; Bretteville, A; Morin, F; Calon, F; Hébert, SS; Whittington, RA; Planel, E
PloS one
9
e94251
2014
Mostra il sommario
Aggregates of hyperphosphorylated tau protein are found in a group of diseases called tauopathies, which includes Alzheimer's disease. The causes and consequences of tau hyperphosphorylation are routinely investigated in laboratory animals. Mice are the models of choice as they are easily amenable to transgenic technology; consequently, their tau phosphorylation levels are frequently monitored by Western blotting using a panel of monoclonal/polyclonal anti-tau antibodies. Given that mouse secondary antibodies can recognize endogenous mouse immunoglobulins (Igs) and the possible lack of specificity with some polyclonal antibodies, non-specific signals are commonly observed. Here, we characterized the profiles of commonly used anti-tau antibodies in four different mouse models: non-transgenic mice, tau knock-out (TKO) mice, 3xTg-AD mice, and hypothermic mice, the latter a positive control for tau hyperphosphorylation. We identified 3 tau monoclonal antibody categories: type 1, characterized by high non-specificity (AT8, AT180, MC1, MC6, TG-3), type 2, demonstrating low non-specificity (AT270, CP13, CP27, Tau12, TG5), and type 3, with no non-specific signal (DA9, PHF-1, Tau1, Tau46). For polyclonal anti-tau antibodies, some displayed non-specificity (pS262, pS409) while others did not (pS199, pT205, pS396, pS404, pS422, A0024). With monoclonal antibodies, most of the interfering signal was due to endogenous Igs and could be eliminated by different techniques: i) using secondary antibodies designed to bind only non-denatured Igs, ii) preparation of a heat-stable fraction, iii) clearing Igs from the homogenates, and iv) using secondary antibodies that only bind the light chain of Igs. All of these techniques removed the non-specific signal; however, the first and the last methods were easier and more reliable. Overall, our study demonstrates a high risk of artefactual signal when performing Western blotting with routinely used anti-tau antibodies, and proposes several solutions to avoid non-specific results. We strongly recommend the use of negative (i.e., TKO) and positive (i.e., hypothermic) controls in all experiments. | | | 24788298
|
AAV-mediated overexpression of neuroserpin in the hippocampus decreases PSD-95 expression but does not affect hippocampal-dependent learning and memory.
Tsang, VW; Young, D; During, MJ; Birch, NP
PloS one
9
e91050
2014
Mostra il sommario
Neuroserpin is a serine protease inhibitor, or serpin, that is expressed in the nervous system and inhibits the protease tissue plasminogen activator (tPA). Neuroserpin has been suggested to play a role in learning and memory but direct evidence for such a role is lacking. Here we have used an adeno-associated virus (AAV) vector expression system to investigate the effect of neuroserpin on hippocampal-dependent learning and memory in the young adult rat. A FLAG-tagged neuroserpin construct was initially characterized by in vitro transcription/translation and transfection into HEK293 cells and shown to interact with tPA and be targeted to the secretory pathway. Targeted injection of a chimeric AAV1/2 vector expressing FLAG-neuroserpin resulted in localized overexpression in the dorsal hippocampus. Neuroserpin overexpression led to the appearance of an unstable neuroserpin:tPA complex in zymographic assays consistent with interaction with endogenous tPA in vivo. Rats overexpressing neuroserpin also showed a significant decrease in the levels of postsynaptic density protein 95, a major postsynaptic scaffolding protein. Three weeks after injection, a range of behavioural tests was performed to measure spatial and associative learning and memory, as well as innate and acquired fear. These tests provided no evidence of a role for neuroserpin in hippocampal-dependent learning and memory. In summary this study does not support a role for neuroserpin in hippocampal-dependent learning and memory in young adult rats but does suggest an involvement of neuroserpin in hippocampal synaptic plasticity. | | | 24608243
|
Improved application of the electrophoretic tissue clearing technology, CLARITY, to intact solid organs including brain, pancreas, liver, kidney, lung, and intestine.
Lee, H; Park, JH; Seo, I; Park, SH; Kim, S
BMC developmental biology
14
48
2014
Mostra il sommario
Mapping of tissue structure at the cellular, circuit, and organ-wide scale is important for understanding physiological and biological functions. A bio-electrochemical technique known as CLARITY used for three-dimensional anatomical and phenotypical mapping within transparent intact tissues has been recently developed. This method provided a major advance in understanding the structure-function relationships in circuits of the nervous system and organs by using whole-body clearing. Thus, in the present study, we aimed to improve the original CLARITY procedure and developed specific CLARITY protocols for various intact organs.We determined the optimal conditions for reducing bubble formation, discoloration, and depositing of black particles on the surface of tissue, which allowed production of clearer organ images. We also determined the appropriate replacement cycles of clearing solution for each type of organ, and convincingly demonstrated that 250-280 mA is the ideal range of electrical current for tissue clearing. We then acquired each type of cleared organs including brain, pancreas, liver, lung, kidney, and intestine. Additionally, we determined the images of axon fibers of hippocampal region, the Purkinje layer of cerebellum, and vessels and cellular nuclei of pancreas.CLARITY is an innovative biochemical technology for the structural and molecular analysis of various types of tissue. We developed improved CLARITY methods for clearing of the brain, pancreas, lung, intestine, liver, and kidney, and identified the appropriate experimental conditions for clearing of each specific tissue type. These optimized methods will be useful for the application of CLARITY to various types of organs. | | | 25528649
|
Structural basis for extracellular cis and trans RPTPσ signal competition in synaptogenesis.
Coles, CH; Mitakidis, N; Zhang, P; Elegheert, J; Lu, W; Stoker, AW; Nakagawa, T; Craig, AM; Jones, EY; Aricescu, AR
Nature communications
5
5209
2014
Mostra il sommario
Receptor protein tyrosine phosphatase sigma (RPTPσ) regulates neuronal extension and acts as a presynaptic nexus for multiple protein and proteoglycan interactions during synaptogenesis. Unknown mechanisms govern the shift in RPTPσ function, from outgrowth promotion to synaptic organization. Here, we report crystallographic, electron microscopic and small-angle X-ray scattering analyses, which reveal sufficient inter-domain flexibility in the RPTPσ extracellular region for interaction with both cis (same cell) and trans (opposite cell) ligands. Crystal structures of RPTPσ bound to its postsynaptic ligand TrkC detail an interaction surface partially overlapping the glycosaminoglycan-binding site. Accordingly, heparan sulphate and heparin oligomers compete with TrkC for RPTPσ binding in vitro and disrupt TrkC-dependent synaptic differentiation in neuronal co-culture assays. We propose that transient RPTPσ ectodomain emergence from the presynaptic proteoglycan layer allows capture by TrkC to form a trans-synaptic complex, the consequent reduction in RPTPσ flexibility potentiating interactions with additional ligands to orchestrate excitatory synapse formation. | | | 25385546
|
MHC class I limits hippocampal synapse density by inhibiting neuronal insulin receptor signaling.
Dixon-Salazar, TJ; Fourgeaud, L; Tyler, CM; Poole, JR; Park, JJ; Boulanger, LM
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
11844-56
2014
Mostra il sommario
Proteins of the major histocompatibility complex class I (MHCI) negatively regulate synapse density in the developing vertebrate brain (Glynn et al., 2011; Elmer et al., 2013; Lee et al., 2014), but the underlying mechanisms remain largely unknown. Here we identify a novel MHCI signaling pathway that involves the inhibition of a known synapse-promoting factor, the insulin receptor. Dominant-negative insulin receptor constructs decrease synapse density in the developing Xenopus visual system (Chiu et al., 2008), and insulin receptor activation increases dendritic spine density in mouse hippocampal neurons in vitro (Lee et al., 2011). We find that genetically reducing cell surface MHCI levels increases synapse density selectively in regions of the hippocampus where insulin receptors are expressed, and occludes the neuronal insulin response by de-repressing insulin receptor signaling. Pharmacologically inhibiting insulin receptor signaling in MHCI-deficient animals rescues synapse density, identifying insulin receptor signaling as a critical mediator of the tonic inhibitory effects of endogenous MHCI on synapse number. Insulin receptors co-immunoprecipitate MHCI from hippocampal lysates, and MHCI unmasks a cytoplasmic epitope of the insulin receptor that mediates downstream signaling. These results identify an important role for an MHCI-insulin receptor signaling pathway in circuit patterning in the developing brain, and suggest that changes in MHCI expression could unexpectedly regulate neuronal insulin sensitivity in the aging and diseased brain. | | | 25164678
|
A novel Hap1-Tsc1 interaction regulates neuronal mTORC1 signaling and morphogenesis in the brain.
Mejia, LA; Litterman, N; Ikeuchi, Y; de la Torre-Ubieta, L; Bennett, EJ; Zhang, C; Harper, JW; Bonni, A
The Journal of neuroscience : the official journal of the Society for Neuroscience
33
18015-21
2013
Mostra il sommario
Tuberous sclerosis complex (TSC) is a leading genetic cause of autism. The TSC proteins Tsc1 and Tsc2 control the mTORC1 signaling pathway in diverse cells, but how the mTORC1 pathway is specifically regulated in neurons remains to be elucidated. Here, using an interaction proteomics approach in neural cells including neurons, we uncover the brain-enriched protein huntingtin-associated protein 1 (Hap1) as a novel functional partner of Tsc1. Knockdown of Hap1 promotes specification of supernumerary axons in primary hippocampal neurons and profoundly impairs the positioning of pyramidal neurons in the mouse hippocampus in vivo. The Hap1 knockdown-induced phenotypes in primary neurons and in vivo recapitulate the phenotypes induced by Tsc1 knockdown. We also find that Hap1 knockdown in hippocampal neurons induces the downregulation of Tsc1 and stimulates the activity of mTORC1, as reflected by phosphorylation of the ribosomal protein S6. Inhibition of mTORC1 activity suppresses the Hap1 knockdown-induced polarity phenotype in hippocampal neurons. Collectively, these findings define a novel link between Hap1 and Tsc1 that regulates neuronal mTORC1 signaling and neuronal morphogenesis, with implications for our understanding of developmental disorders of cognition. | | | 24227713
|
Interaction of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival.
Zurashvili, T; Cordón-Barris, L; Ruiz-Babot, G; Zhou, X; Lizcano, JM; Gómez, N; Giménez-Llort, L; Bayascas, JR
Molecular and cellular biology
33
1027-40
2013
Mostra il sommario
3-Phosphoinositide-dependent protein kinase 1 (PDK1) operates in cells in response to phosphoinositide 3-kinase activation and phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P(3)] production by activating a number of AGC kinases, including protein kinase B (PKB)/Akt. Both PDK1 and PKB contain pleckstrin homology (PH) domains that interact with the PtdIns(3,4,5)P(3) second messenger. Disrupting the interaction of the PDK1 PH domain with phosphoinositides by expressing the PDK1 K465E knock-in mutation resulted in mice with reduced PKB activation. We explored the physiological consequences of this biochemical lesion in the central nervous system. The PDK1 knock-in mice displayed a reduced brain size due to a reduction in neuronal cell size rather than cell number. Reduced BDNF-induced phosphorylation of PKB at Thr308, the PDK1 site, was observed in the mutant neurons, which was not rate limiting for the phosphorylation of those PKB substrates governing neuronal survival and apoptosis, such as FOXO1 or glycogen synthase kinase 3 (GSK3). Accordingly, the integrity of the PDK1 PH domain was not essential to support the survival of different embryonic neuronal populations analyzed. In contrast, PKB-mediated phosphorylation of PRAS40 and TSC2, allowing optimal mTORC1 activation and brain-specific kinase (BRSK) protein synthesis, was markedly reduced in the mutant mice, leading to impaired neuronal growth and differentiation. | Immunofluorescence | | 23275438
|