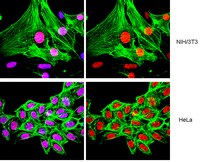
Class I histone deacetylase inhibitors inhibit the retention of BRCA1 and 53BP1 at the site of DNA damage.
Fukuda, T; Wu, W; Okada, M; Maeda, I; Kojima, Y; Hayami, R; Miyoshi, Y; Tsugawa, K; Ohta, T
Cancer science
106
1050-6
2015
Show Abstract
BRCA1 and 53BP1 antagonistically regulate homology-directed repair (HDR) and non-homologous end-joining (NHEJ) of DNA double-strand breaks (DSB). The histone deacetylase (HDAC) inhibitor trichostatin A directly inhibits the retention of 53BP1 at DSB sites by acetylating histone H4 (H4ac), which interferes with 53BP1 binding to dimethylated histone H4 Lys20 (H4K20me2). Conversely, we recently found that the retention of the BRCA1/BARD1 complex is also affected by another methylated histone residue, H3K9me2, which can be suppressed by the histone lysine methyltransferase (HKMT) inhibitor UNC0638. Here, we investigate the effects of the class I HDAC inhibitors MS-275 and FK228 compared to UNC0638 on histone modifications and the DNA damage response. In addition to H4ac, the HDAC inhibitors induce H3K9ac and inhibit H3K9me2 at doses that do not affect the expression levels of DNA repair genes. By contrast, UNC0638 selectively inhibits H3K9me2 without affecting the levels of H3K9ac, H3K56ac or H4ac. Reflecting their effects on histone modifications, the HDAC inhibitors inhibit ionizing radiation-induced foci (IRIF) formation of BRCA1 and BARD1 as well as 53BP1 and RIF1, whereas UNC0638 suppresses IRIF formation of BRCA1 and BARD1 but not 53BP1 and RIF1. Although HDAC inhibitors suppressed HDR, they did not cooperate with the poly(ADP-ribose) polymerase inhibitor olaparib to block cancer cell growth, possibly due to simultaneous suppression of NHEJ pathway components. Collectively, these results suggest the mechanism by that HDAC inhibitors inhibit both the HDR and NHEJ pathways, whereas HKMT inhibitor inhibits only the HDR pathway; this finding may affect the chemosensitizing effects of the inhibitors. | | | 26053117
 |
Epigenetic memory gained by priming with osteogenic induction medium improves osteogenesis and other properties of mesenchymal stem cells.
Rui, Y; Xu, L; Chen, R; Zhang, T; Lin, S; Hou, Y; Liu, Y; Meng, F; Liu, Z; Ni, M; Tsang, KS; Yang, F; Wang, C; Chan, HC; Jiang, X; Li, G
Scientific reports
5
11056
2015
Show Abstract
Mesenchymal stem cells (MSCs) are highly plastic cells that are able to transdifferentiate or dedifferentiate under appropriate conditions. In the present study, we reported here that after in vitro induction of osteogenic differentiation, MSCs could be reverted to a primitive stem cell population (dedifferentiated osteogenic MSCs, De-Os-MSCs) with improved cell survival, colony formation, osteogenic potential, migratory capacity and increased expression of Nanog, Oct4 and Sox2. Most importantly, our results showed great superiority of the De-Os-MSCs over untreated MSCs in ectopic bone formation in vivo. Furthermore, Nanog-knockdown in MSCs could reverse these enhanced properties in De-Os-MSCs in vitro, indicating a central role of Nanog in the transcriptional network. In addition, epigenetic regulations including DNA methylation and histone modifications may play important roles in regulating the de-osteogenic differentiation process. And we found decreased methylation and promoter accrual of activating histone marks, such as H3K4me3 and H4ac on both Nanog and Oct4 gene promoters. Taken together, our study demonstrated that epigenetic memory in De-Os-MSCs gained by priming with osteogenic induction medium favored their differentiation along osteoblastic lineage with improved cell survival and migratory abilities, which may have application potential in enhancing their regenerative capacity in mammals. | | | 26053250
|
SCARECROW-LIKE15 interacts with HISTONE DEACETYLASE19 and is essential for repressing the seed maturation programme.
Gao, MJ; Li, X; Huang, J; Gropp, GM; Gjetvaj, B; Lindsay, DL; Wei, S; Coutu, C; Chen, Z; Wan, XC; Hannoufa, A; Lydiate, DJ; Gruber, MY; Chen, ZJ; Hegedus, DD
Nature communications
6
7243
2015
Show Abstract
Epigenetic regulation of gene expression is critical for controlling embryonic properties during the embryo-to-seedling phase transition. Here we report that a histone deacetylase19 (HDA19)-associated regulator, scarecrow-like15 (SCL15), is essential for repressing the seed maturation programme in vegetative tissues. SCL15 is expressed in and GFP-tagged SCL15 predominantly localizes to, the vascular bundles particularly in the phloem companion cells and neighbouring specialized cells. Mutation of SCL15 leads to a global shift in gene expression in seedlings to a profile resembling late embryogenesis in seeds. In scl15 seedlings, many genes involved in seed maturation are markedly derepressed with concomitant accumulation of seed 12S globulin; this is correlated with elevated levels of histone acetylation at a subset of seed-specific loci. SCL15 physically interacts with HDA19 and direct targets of HDA19-SCL15 association are identified. These studies reveal that SCL15 acts as an HDA19-associated regulator to repress embryonic traits in seedlings. | | | 26129778
|
HDAC8, A Potential Therapeutic Target for the Treatment of Malignant Peripheral Nerve Sheath Tumors (MPNST).
Lopez, G; Bill, KL; Bid, HK; Braggio, D; Constantino, D; Prudner, B; Zewdu, A; Batte, K; Lev, D; Pollock, RE
PloS one
10
e0133302
2015
Show Abstract
HDAC isoform-specific inhibitors may improve the therapeutic window while limiting toxicities. Developing inhibitors against class I isoforms poses difficulties as they share high homology among their catalytic sites; however, HDAC8 is structurally unique compared to other class I isoforms. HDAC8 inhibitors are novel compounds and have affinity for class I HDAC isoforms demonstrating anti-cancer effects; little is known about their activity in malignant peripheral nerve sheath tumors (MPNST). Recently, we demonstrated anti-MPNST efficacy of HDAC8i in human and murine-derived MPNST pre-clinical models; we now seek to consider the potential therapeutic inhibition of HDAC8 in MPNST.Four Human MPNST cell lines, a murine-derived MPNST cell line, and two HDAC8 inhibitors (PCI-34051, PCI-48012; Pharmacyclics, Inc. Sunnyvale, CA) were studied. Proliferation was determined using MTS and clonogenic assays. Effects on cell cycle were determined via PI FACS analysis; effects on apoptosis were determined using Annexin V-PI FACS analysis and cleaved caspase 3 expression. In vivo growth effects of HDAC8i were evaluated using MPNST xenograft models. 2D gel electrophoresis and mass spectrometry were used to identify potential HDAC8 deacetylation substrates.HDAC8i induced cell growth inhibition and marked S-phase cell cycle arrest in human and murine-derived MPNST cells. Relative to control, HDAC8i induced apoptosis in both human and murine-derived MPNST cells. HDAC8i exhibited significant effects on MPNST xenograft growth (p=0.001) and tumor weight (p=0.02). Four potential HDAC8 substrate targets were identified using a proteomic approach: PARK7, HMGB1, PGAM1, PRDX6.MPNST is an aggressive sarcoma that is notoriously therapy-resistant, hence the urgent need for improved anti-MPNST therapies. HDAC8 inhibition may be useful for MPNST by improving efficacy while limiting toxicities as compared to pan-HDACis. | | | 26200462
|
Epigenetic synergy between decitabine and platinum derivatives.
Qin, T; Si, J; Raynal, NJ; Wang, X; Gharibyan, V; Ahmed, S; Hu, X; Jin, C; Lu, Y; Shu, J; Estecio, MR; Jelinek, J; Issa, JP
Clinical epigenetics
7
97
2015
Show Abstract
Aberrant epigenetic silencing of tumor suppressor genes has been recognized as a driving force in cancer. Epigenetic drugs such as the DNA methylation inhibitor decitabine reactivate genes and are effective in myeloid leukemia, but resistance often develops and efficacy in solid tumors is limited. To improve their clinical efficacy, we searched among approved anti-cancer drugs for an epigenetic synergistic combination with decitabine.We used the YB5 cell line, a clonal derivative of the SW48 colon cancer cell line that contains a single copy of a hypermethylated cytomegalovirus (CMV) promoter driving green fluorescent protein (GFP) to screen for drug-induced gene reactivation and synergy with decitabine. None of the 16 anti-cancer drugs tested had effects on their own. However, in combination with decitabine, platinum compounds showed striking synergy in activating GFP. This was dose dependent, observed both in concurrent and sequential combinations, and also seen with other alkylating agents. Clinically achievable concentrations of carboplatin at (25 μM) and decitabine reactivated GFP in 28 % of the YB5 cells as compared to 15 % with decitabine alone. Epigenetic synergy was also seen at endogenously hypermethylated tumor suppressor genes such as MLH1 and PDLIM4. Genome-wide studies showed that reactivation of hypermethylated genes by the combination was significantly better than that induced by decitabine alone or carboplatin alone. Platinum compounds did not enhance decitabine-induced hypomethylation. Rather, we found significantly inhibited HP1α expression by carboplatin and the combination. This was accompanied by increased histone H3 lysine 4 (H3K4) trimethylation and histone H3 lysine 9 (H3K9) acetylation at reactivated genes (P less than 0.0001) and reduced occupancy by methyl-binding proteins including MeCP2 and methyl-CpG-binding domain protein 2 (MBD2) (P less than 0.0001).Our results suggest that the combination of decitabine with platinum analogs shows epigenetic synergy that might be exploited in the treatment of different cancers. | | | 26366234
|
A comprehensive epigenome map of Plasmodium falciparum reveals unique mechanisms of transcriptional regulation and identifies H3K36me2 as a global mark of gene suppression.
Karmodiya, K; Pradhan, SJ; Joshi, B; Jangid, R; Reddy, PC; Galande, S
Epigenetics & chromatin
8
32
2015
Show Abstract
Role of epigenetic mechanisms towards regulation of the complex life cycle/pathogenesis of Plasmodium falciparum, the causative agent of malaria, has been poorly understood. To elucidate stage-specific epigenetic regulation, we performed genome-wide mapping of multiple histone modifications of P. falciparum. Further to understand the differences in transcription regulation in P. falciparum and its host, human, we compared their histone modification profiles.Our comprehensive comparative analysis suggests distinct mode of transcriptional regulation in malaria parasite by virtue of poised genes and differential histone modifications. Furthermore, analysis of histone modification profiles predicted 562 genes producing anti-sense RNAs and 335 genes having bidirectional promoter activity, which raises the intriguing possibility of RNA-mediated regulation of transcription in P. falciparum. Interestingly, we found that H3K36me2 acts as a global repressive mark and gene regulation is fine tuned by the ratio of activation marks to H3K36me2 in P. falciparum. This novel mechanism of gene regulation is supported by the fact that knockout of SET genes (responsible for H3K36 methylation) leads to up-regulation of genes with highest occupancy of H3K36me2 in wild-type P. falciparum. Moreover, virulence (var) genes are mostly poised and marked by a unique set of activation (H4ac) and repression (H3K9me3) marks, which are mutually exclusive to other Plasmodium housekeeping genes.Our study reveals unique plasticity in the epigenetic regulation in P. falciparum which can influence parasite virulence and pathogenicity. The observed differences in the histone code and transcriptional regulation in P. falciparum and its host will open new avenues for epigenetic drug development against malaria parasite. | | | 26388940
|
Nanocurcumin Prevents Hypoxia Induced Stress in Primary Human Ventricular Cardiomyocytes by Maintaining Mitochondrial Homeostasis.
Nehra, S; Bhardwaj, V; Ganju, L; Saraswat, D
PloS one
10
e0139121
2015
Show Abstract
Hypoxia induced oxidative stress incurs pathophysiological changes in hypertrophied cardiomyocytes by promoting translocation of p53 to mitochondria. Here, we investigate the cardio-protective efficacy of nanocurcumin in protecting primary human ventricular cardiomyocytes (HVCM) from hypoxia induced damages. Hypoxia induced hypertrophy was confirmed by FITC-phenylalanine uptake assay, atrial natriuretic factor (ANF) levels and cell size measurements. Hypoxia induced translocation of p53 was investigated by using mitochondrial membrane permeability transition pore blocker cyclosporin A (blocks entry of p53 to mitochondria) and confirmed by western blot and immunofluorescence. Mitochondrial damage in hypertrophied HVCM cells was evaluated by analysing bio-energetic, anti-oxidant and metabolic function and substrate switching form lipids to glucose. Nanocurcumin prevented translocation of p53 to mitochondria by stabilizing mitochondrial membrane potential and de-stressed hypertrophied HVCM cells by significant restoration in lactate, acetyl-coenzyme A, pyruvate and glucose content along with lactate dehydrogenase (LDH) and 5' adenosine monophosphate-activated protein kinase (AMPKα) activity. Significant restoration in glucose and modulation of GLUT-1 and GLUT-4 levels confirmed that nanocurcumin mediated prevention of substrate switching. Nanocurcumin prevented of mitochondrial stress as confirmed by c-fos/c-jun/p53 signalling. The data indicates decrease in p-300 histone acetyl transferase (HAT) mediated histone acetylation and GATA-4 activation as pharmacological targets of nanocurcumin in preventing hypoxia induced hypertrophy. The study provides an insight into propitious therapeutic effects of nanocurcumin in cardio-protection and usability in clinical applications. | | | 26406246
|
Transcriptional regulation of the human TNFSF11 gene in T cells via a cell type-selective set of distal enhancers.
Bishop, KA; Wang, X; Coy, HM; Meyer, MB; Gumperz, JE; Pike, JW
Journal of cellular biochemistry
116
320-30
2015
Show Abstract
In addition to osteoblast lineage cells, the TNF-like factor receptor activator of NF-κB ligand (RANKL) is expressed in both B and T cells and may play a role in bone resorption. Rankl gene (Tnfsf11) expression in mouse T cells is mediated through multiple distal elements marked by increased transcription factor occupancy, histone tail acetylation, and RNA polymerase II recruitment. Little is known, however, of the regulation of human TNFSF11 in T cells. Accordingly, we examined the consequence of T cell activation on the expression of this factor both in Jurkat cells and in primary human T cells. We then explored the mechanism of this regulation by scanning over 400 kb of DNA surrounding the TNFSF11 locus for regulatory enhancers using ChIP-chip analysis. Histone H3/H4 acetylation enrichment identified putative regulatory regions located between -170 and -220 kb upstream of the human TNFSF11 TSS that we designated the human T cell control region (hTCCR). This region showed high sequence conservation with the mouse TCCR. Inhibition of MEK1/2 by U0126 resulted in decreased RANKL expression suggesting that stimulation through MEK1/2 was a prerequisite. ChIP-chip analysis also revealed that c-FOS was recruited to the hTCCR as well. Importantly, both the human TNFSF11 D5a/b (RLD5a/b) enhancer and segments of the hTCCR mediated robust inducible reporter activity following TCR activation. Finally, SNPs implicated in diseases characterized by dysregulated BMD co-localized to the hTCCR region. We conclude that the hTCCR region contains a cell-selective set of enhancers that plays an integral role in the transcriptional regulation of the TNFSF11 gene in human T cells. | | | 25211367
|
Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcriptional activation.
Wang, R; You, J
The Journal of biological chemistry
290
2744-58
2015
Show Abstract
NUT midline carcinoma (NMC) is a rare but highly aggressive cancer typically caused by the translocation t(15;19), which results in the formation of the BRD4-NUT fusion oncoprotein. Previous studies have demonstrated that fusion of the NUT protein with the double bromodomains of BRD4 may significantly alter the cellular gene expression profile to contribute to NMC tumorigenesis. However, the mechanistic details of this BRD4-NUT function remain poorly understood. In this study, we examined the NUT function in transcriptional regulation by targeting it to a LacO transgene array integrated in U2OS 2-6-3 cells, which allow us to visualize how NUT alters the in situ gene transcription dynamic. Using this system, we demonstrated that the NUT protein tethered to the LacO locus recruits p300/CREB-binding protein (CBP), induces histone hyperacetylation, and enriches BRD4 to the transgene array chromatin foci. We also discovered that, in BRD4-NUT expressed in NMC cells, the NUT moiety of the fusion protein anchored to chromatin by the double bromodomains also stimulates histone hyperacetylation, which causes BRD4 to bind tighter to chromatin. Consequently, multiple BRD4-interacting factors are recruited to the NUT-associated chromatin locus to activate in situ transgene expression. This gene transcription function was repressed by either expression of a dominant negative inhibitor of the p300-NUT interaction or treatment with (+)-JQ1, which dissociates BRD4 from the LacO chromatin locus. Our data support a model in which BRD4-NUT-stimulated histone hyperacetylation recruits additional BRD4 and interacting partners to support transcriptional activation, which underlies the BRD4-NUT oncogenic mechanism in NMC. | Immunofluorescence | | 25512383
|
Loss of the Notch effector RBPJ promotes tumorigenesis.
Kulic, I; Robertson, G; Chang, L; Baker, JH; Lockwood, WW; Mok, W; Fuller, M; Fournier, M; Wong, N; Chou, V; Robinson, MD; Chun, HJ; Gilks, B; Kempkes, B; Thomson, TA; Hirst, M; Minchinton, AI; Lam, WL; Jones, S; Marra, M; Karsan, A
The Journal of experimental medicine
212
37-52
2015
Show Abstract
Aberrant Notch activity is oncogenic in several malignancies, but it is unclear how expression or function of downstream elements in the Notch pathway affects tumor growth. Transcriptional regulation by Notch is dependent on interaction with the DNA-binding transcriptional repressor, RBPJ, and consequent derepression or activation of associated gene promoters. We show here that RBPJ is frequently depleted in human tumors. Depletion of RBPJ in human cancer cell lines xenografted into immunodeficient mice resulted in activation of canonical Notch target genes, and accelerated tumor growth secondary to reduced cell death. Global analysis of activated regions of the genome, as defined by differential acetylation of histone H4 (H4ac), revealed that the cell death pathway was significantly dysregulated in RBPJ-depleted tumors. Analysis of transcription factor binding data identified several transcriptional activators that bind promoters with differential H4ac in RBPJ-depleted cells. Functional studies demonstrated that NF-κB and MYC were essential for survival of RBPJ-depleted cells. Thus, loss of RBPJ derepresses target gene promoters, allowing Notch-independent activation by alternate transcription factors that promote tumorigenesis. | | | 25512468
|