Wenn Sie das Fenster schließen, wird Ihre Konfiguration nicht gespeichert, es sei denn, Sie haben Ihren Artikel in die Bestellung aufgenommen oder zu Ihren Favoriten hinzugefügt.
Klicken Sie auf OK, um das MILLIPLEX® MAP-Tool zu schließen oder auf Abbrechen, um zu Ihrer Auswahl zurückzukehren.
Wählen Sie konfigurierbare Panels & Premixed-Kits - ODER - Kits für die zelluläre Signaltransduktion & MAPmates™
Konfigurieren Sie Ihre MILLIPLEX® MAP-Kits und lassen sich den Preis anzeigen.
Konfigurierbare Panels & Premixed-Kits
Unser breites Angebot enthält Multiplex-Panels, für die Sie die Analyten auswählen können, die am besten für Ihre Anwendung geeignet sind. Unter einem separaten Register können Sie das Premixed-Cytokin-Format oder ein Singleplex-Kit wählen.
Kits für die zelluläre Signaltransduktion & MAPmates™
Wählen Sie gebrauchsfertige Kits zur Erforschung gesamter Signalwege oder Prozesse. Oder konfigurieren Sie Ihre eigenen Kits mit Singleplex MAPmates™.
Die folgenden MAPmates™ sollten nicht zusammen analysiert werden: -MAPmates™, die einen unterschiedlichen Assaypuffer erfordern. -Phosphospezifische und MAPmate™ Gesamtkombinationen wie Gesamt-GSK3β und Gesamt-GSK3β (Ser 9). -PanTyr und locusspezifische MAPmates™, z.B. Phospho-EGF-Rezeptor und Phospho-STAT1 (Tyr701). -Mehr als 1 Phospho-MAPmate™ für ein einziges Target (Akt, STAT3). -GAPDH und β-Tubulin können nicht mit Kits oder MAPmates™, die panTyr enthalten, analysiert werden.
.
Bestellnummer
Bestellinformationen
St./Pkg.
Liste
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Wählen Sie bitte Spezies, Panelart, Kit oder Probenart
Um Ihr MILLIPLEX® MAP-Kit zu konfigurieren, wählen Sie zunächst eine Spezies, eine Panelart und/oder ein Kit.
Custom Premix Selecting "Custom Premix" option means that all of the beads you have chosen will be premixed in manufacturing before the kit is sent to you.
Catalogue Number
Ordering Description
Qty/Pack
List
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Spezies
Panelart
Gewähltes Kit
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
96-Well Plate
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
Weitere Reagenzien hinzufügen (MAPmates erfordern die Verwendung eines Puffer- und Detektionskits)
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
48-602MAG
Buffer Detection Kit for Magnetic Beads
1 Kit
Platzsparende Option Kunden, die mehrere Kits kaufen, können ihre Multiplex-Assaykomponenten in Kunststoffbeuteln anstelle von Packungen erhalten, um eine kompaktere Lagerung zu ermöglichen.
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Das Produkt wurde in Ihre Bestellung aufgenommen
Sie können nun ein weiteres Kit konfigurieren, ein Premixed-Kit wählen, zur Kasse gehen oder das Bestell-Tool schließen.
This gene encodes the alpha-1 chain of type II collagen, a fibrillar collagen found in cartilage and the vitreous humor of the eye. Mutations in this gene are associated with achondrogenesis, chondrodysplasia, early onset familial osteoarthritis, SED congenita, Langer-Saldino achondrogenesis, Kniest dysplasia, Stickler syndrome type I, and spondyloepimetaphyseal dysplasia Strudwick type. In addition, defects in processing chondrocalcin, a calcium binding protein that is the C-propeptide of this collagen molecule, are also associated with chondrodysplasia. There are two transcripts identified for this gene.
FUNCTION: SwissProt: P02458 # Type II collagen is specific for cartilaginous tissues. It is essential for the normal embryonic development of the skeleton, for linear growth and for the ability of cartilage to resist compressive forces. SIZE: 1487 amino acids; 141785 Da SUBUNIT: Homotrimers of alpha 1(II) chains. SUBCELLULAR LOCATION: Secreted, extracellular space, extracellular matrix (By similarity). TISSUE SPECIFICITY: High expression of isoform 2 in juvenile chondrocyte and low in fetal chondrocyte. PTM: Prolines at the third position of the tripeptide repeating unit (G-X-Y) are hydroxylated in some or all of the chains. & The N-telopeptide is covalently linked to the helical COL2 region of alpha 1(IX), alpha 2(IX) and alpha 3(IX) chain. The C- telopeptide is covalently linked to an another site in the helical region of alpha 3(IX) COL2. DISEASE: SwissProt: P02458 # Defects in COL2A1 are the cause of a variety of chondrodysplasia including hypochondrogenesis and osteoarthritis. & Defects in COL2A1 are the cause of spondyloepiphyseal dysplasia congenital type (SEDC) [MIM:183900]. This disorder is characterized by disproportionate short stature and pleiotropic involvement of the skeletal and ocular systems. & Defects in COL2A1 are the cause of Strudwick type spondyloepimetaphyseal dysplasia (SEMD) [MIM:184250]. SEMD is characterized by disproportionate short stature, pectus carinatum, and scoliosis, as well as dappled metaphyses (which is not seen in SEDC). & Defects in COL2A1 are the cause of achondrogenesis hypochondrogenesis type 2 (ACG2) [MIM:200610]. ACG2 is a disease characterized by the absence of ossification in the vertebral column, sacrum and pubic bones. & Defects in COL2A1 are the cause of Legg-Calve-Perthes disease (LCPD) [MIM:150600]; also known as Legg-Perthes disease or Perthes disease. LCPD is characterized by loss of circulation to the femoral head, resulting in avascular necrosis in a growing child. Clinical pictures of the disease vary, depending on the phase of disease progression through ischemia, revascularization, fracture and collapse, and repair and remodeling of the bone. & Defects in COL2A1 are the cause of Kniest syndrome (KS) [MIM:156550]; also known as Kniest dysplasia or metatropic dwarfism type II. KS is a moderately severe chondrodysplasia phenotype that results from mutations in the COL2A1 gene. Characteristics of the disorder include a short trunk and extremities, mid-face hypoplasia, cleft palate, myopia, retinal detachment, and hearing loss. & Defects in COL2A1 are a cause of primary avascular necrosis of femoral head (ANFH) [MIM:608805]; also called ischemic necrosis of the femoral head or osteonecrosis of the femoral head. ANFH causes disability that often requires surgical intervention. Most cases are sporadic, but families in which there is an autosomal dominant inheritance of the disease have been identified. It has been estimated that 300,000 to 600,000 people in the United States have ANFH. Approximately 15,000 new cases of this common and disabling disorder are reported annually. The age at the onset is earlier than that for osteoarthritis. The diagnosis is typically made when patients are between the ages of 30 and 60 years. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. Moreover, nearly 10 percent of the 500,000 total-hip arthroplasties performed each year in the United States involve patients with ANFH. As a result, this disease creates a substantial socioeconomic cost as well as a burden for patients and their families. & Defects in COL2A1 are the cause of osteoarthritis with mild chondrodysplasia [MIM:604864]. Osteoarthritis is a common disease that produces joint pain and stiffness together with radiologic evidence of progressive degeneration of joint cartilage. Some forms of osteoarthritis are secondary to events such as trauma, infections, metabolic disorders, or congenital or heritable conditions that deform the epiphyses or related structures. In most patients, however, there is no readily identifiable cause of osteoarthritis. Inheritance in a Mendelian dominant manner has been demonstrated in some families with primary generalized osteoarthritis. Reports demonstrate coinheritance of primary generalized osteoarthritis with specific alleles of the gene COL2A1, the precursor of the major protein of cartilage. & Defects in COL2A1 are the cause of platyspondylic lethal skeletal dysplasia Torrance type (PLSD-T) [MIM:151210]. Platyspondylic lethal skeletal dysplasias (PLSDs) are a heterogeneous group of chondrodysplasias characterized by severe platyspondyly and limb shortening. PLSD-T is characterized by varying platyspondyly, short ribs with anterior cupping, hypoplasia of the lower ilia with broad ischial and pubic bones, and shortening of the tubular bones with splayed and cupped metaphyses. Histology of the growth plate typically shows focal hypercellularity with slightly enlarged chondrocytes in the resting cartilage and relatively well-preserved columnar formation and ossification at the chondro-osseous junction. PLSD-T is generally a perinatally lethal disease, but a few long-term survivors have been reported. & Defects in COL2A1 are the cause of multiple epiphyseal dysplasia with myopia and conductive deafness (EDMMD) [MIM:132450]. Multiple epiphyseal dysplasia is a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. EDMMD is an autosomal dominant disorder characterized by epiphyseal dysplasia associated with progressive myopia, retinal thinning, crenated cataracts, conductive deafness. & Defects in COL2A1 are the cause of spondyloperipheral dysplasia (SPD) [MIM:271700]. SPD patients manifest short stature, midface hypoplasia, sensorineural hearing loss, spondyloepiphyseal dysplasia, platyspondyly and brachydactyly. & Defects in COL2A1 are the cause of Wagner syndrome type II (WS-II); a disease characterized by early-onset cataracts, lattice degeneration of the retina, and retinal detachment without involvement of monocular tissues. & Defects in COL2A1 are the cause of Stickler syndrome type 1 (STL1) [MIM:108300]; also known as vitreous type 1, or membranous vitreous type. STL1 is an autosomal dominant disorder characterized by progressive myopia beginning in the first decade of life, vitreo-retinal degeneration, retinal detachment, cleft palate, midfacial hypoplasia, osteoarthritis, and sensorineural hearing loss. & Defects in COL2A1 are a cause of autosomal dominant rhegmatogenous retinal detachment (DRRD) [MIM:609508]. RDD most frequently results from retinal tearing at the time of posterior vitreous detachment. Non-syndromic RRD can be inherited in a clearly dominant fashion, although in most of these cases, the genetic locus for the disorder is unknown. However, RRD is also a common feature of the type II collagenopathies (disorders due to mutations in the gene COL2A1) and some recent examples of mutations in this gene suggest that COL2A1 should be considered a candidate gene for dominant RRD (DRRD). & Of special interest are three different variants that replace arginine codons at positions 275, 719 and 989 in the triple-helical domain with codons for cysteine, an amino acid not normally found in the triple-helical domain of type II collagen from any species. They are of special interest, because they are the only amino acid substitutions in the triple-helical domain that replaces a Y-position amino acid and cause a disease phenotype. Also, they are recurrent in that they have been found in more than one unrelated individual. SIMILARITY: SwissProt: P02458 ## Belongs to the fibrillar collagen family. & Contains 1 VWFC domain.
Accuracy
97–112%
Physicochemical Information
Sensitivity
39 ng/mL
Linearity of Dilution
91–109%
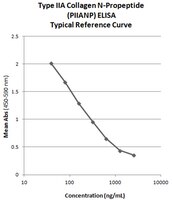
Standard Curve Range
lot dependent
Dimensions
Materials Information
Toxicological Information
Safety Information according to GHS
Safety Information
Product Usage Statements
Usage Statement
For research use only. Not for use in diagnostic procedures.
Tissue structure modification in knee osteoarthritis by use of joint distraction: an open 1-year pilot study. Femke Intema,Peter M Van Roermund,Anne C A Marijnissen,Sebastian Cotofana,Felix Eckstein,Rene M Castelein,Johannes W J Bijlsma,Simon C Mastbergen,Floris P J G Lafeber Annals of the rheumatic diseases
70
2010
Modification of joint tissue damage is challenging in late-stage osteoarthritis (OA). Few options are available for treating end-stage knee OA other than joint replacement.
Millipore offer Collagen antibodies, proteins and kits for your ECM and adhesion research. See below for a list of Collagen related products, based on the expertise of Upstate & Chemicon. Weitere Informationen >>