Wenn Sie das Fenster schließen, wird Ihre Konfiguration nicht gespeichert, es sei denn, Sie haben Ihren Artikel in die Bestellung aufgenommen oder zu Ihren Favoriten hinzugefügt.
Klicken Sie auf OK, um das MILLIPLEX® MAP-Tool zu schließen oder auf Abbrechen, um zu Ihrer Auswahl zurückzukehren.
Wählen Sie konfigurierbare Panels & Premixed-Kits - ODER - Kits für die zelluläre Signaltransduktion & MAPmates™
Konfigurieren Sie Ihre MILLIPLEX® MAP-Kits und lassen sich den Preis anzeigen.
Konfigurierbare Panels & Premixed-Kits
Unser breites Angebot enthält Multiplex-Panels, für die Sie die Analyten auswählen können, die am besten für Ihre Anwendung geeignet sind. Unter einem separaten Register können Sie das Premixed-Cytokin-Format oder ein Singleplex-Kit wählen.
Kits für die zelluläre Signaltransduktion & MAPmates™
Wählen Sie gebrauchsfertige Kits zur Erforschung gesamter Signalwege oder Prozesse. Oder konfigurieren Sie Ihre eigenen Kits mit Singleplex MAPmates™.
Die folgenden MAPmates™ sollten nicht zusammen analysiert werden: -MAPmates™, die einen unterschiedlichen Assaypuffer erfordern. -Phosphospezifische und MAPmate™ Gesamtkombinationen wie Gesamt-GSK3β und Gesamt-GSK3β (Ser 9). -PanTyr und locusspezifische MAPmates™, z.B. Phospho-EGF-Rezeptor und Phospho-STAT1 (Tyr701). -Mehr als 1 Phospho-MAPmate™ für ein einziges Target (Akt, STAT3). -GAPDH und β-Tubulin können nicht mit Kits oder MAPmates™, die panTyr enthalten, analysiert werden.
.
Bestellnummer
Bestellinformationen
St./Pkg.
Liste
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Wählen Sie bitte Spezies, Panelart, Kit oder Probenart
Um Ihr MILLIPLEX® MAP-Kit zu konfigurieren, wählen Sie zunächst eine Spezies, eine Panelart und/oder ein Kit.
Custom Premix Selecting "Custom Premix" option means that all of the beads you have chosen will be premixed in manufacturing before the kit is sent to you.
Catalogue Number
Ordering Description
Qty/Pack
List
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Spezies
Panelart
Gewähltes Kit
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
96-Well Plate
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
Weitere Reagenzien hinzufügen (MAPmates erfordern die Verwendung eines Puffer- und Detektionskits)
Menge
Bestellnummer
Bestellinformationen
St./Pkg.
Listenpreis
48-602MAG
Buffer Detection Kit for Magnetic Beads
1 Kit
Platzsparende Option Kunden, die mehrere Kits kaufen, können ihre Multiplex-Assaykomponenten in Kunststoffbeuteln anstelle von Packungen erhalten, um eine kompaktere Lagerung zu ermöglichen.
Dieser Artikel wurde zu Ihren Favoriten hinzugefügt.
Das Produkt wurde in Ihre Bestellung aufgenommen
Sie können nun ein weiteres Kit konfigurieren, ein Premixed-Kit wählen, zur Kasse gehen oder das Bestell-Tool schließen.
Use Anti-SMN Antibody, clone 62E7 (Mouse Monoclonal Antibody) validated in WB, ICC, IP to detect SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease).
More>>Use Anti-SMN Antibody, clone 62E7 (Mouse Monoclonal Antibody) validated in WB, ICC, IP to detect SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease). Less<<
Anti-SMN Antibody, clone 62E7: SDB (Sicherheitsdatenblätter), Analysenzertifikate und Qualitätszertifikate, Dossiers, Broschüren und andere verfügbare Dokumente.
The survival of motor neurons (SMN) protein is essential for the biogenesis of small nuclear RNA (snRNA)-ribonucleoproteins (snRNPs), the major components of the pre-mRNA splicing machinery. Though it is ubiquitously expressed, SMN deficiency causes the motor neuron degenerative disease spinal muscular atrophy (SMA). SMN deficiency, similar to that which occurs in severe SMA, has unexpected cell type-specific effects on the repertoire of snRNAs and mRNAs. It alters the stoichiometry of snRNAs and causes widespread pre-mRNA splicing defects in numerous transcripts of diverse genes, preferentially those containing a large number of introns, in SMN-deficient mouse tissues. These findings reveal a key role for the SMN complex in RNA metabolism and in splicing regulation and indicate that SMA is a general splicing disease that is not restricted to motor neurons.
References
Product Information
Format
Purified
Control
HeLa cell lysate
Presentation
Purified mouse monoclonal IgG1κ in buffer containing 0.1 M Tris-Glycine (pH 7.4, 150 mM NaCl) with 0.05% sodium azide.
Use Anti-SMN Antibody, clone 62E7 (Mouse Monoclonal Antibody) validated in WB, ICC, IP to detect SMN also known as Spinal muscular atrophy (Werdnig-Hoffmann disease Kugelberg-Welander disease).
Key Applications
Western Blotting
Immunocytochemistry
Immunoprecipitation
Application Notes
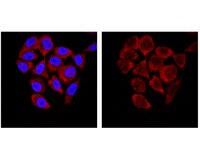
Immunocytochemistry Analysis: 1:500 dilution from a representative lot detected SMN in HeLa cells.
Biological Information
Immunogen
His-tagged recombinant protein corresponding to human SMN.
Epitope
Unknown
Clone
62E7
Concentration
Please refer to the Certificate of Analysis for the lot-specific concentration.
Host
Mouse
Specificity
Recognizes SMN.
Isotype
IgG1κ
Species Reactivity
Human
Mouse
Xenopus
Species Reactivity Note
Demonstrated to react with human. Predicted to react with mouse and xenopus based on sequence homology.
This gene is part of a 500 kb inverted duplication on chromosome 5q13. This duplicated region contains at least four genes and repetitive elements which make it prone to rearrangements and deletions. The repetitiveness and complexity of the sequence have also caused difficulty in determining the organization of this genomic region. The telomeric and centromeric copies of this gene are nearly identical and encode the same protein. However, mutations in this gene, the telomeric copy, are associated with spinal muscular atrophy; mutations in the centromeric copy do not lead to disease. The centromeric copy may be a modifier of disease caused by mutation in the telomeric copy. The critical sequence difference between the two genes is a single nucleotide in exon 7, which is thought to be an exon splice enhancer. Note that the nine exons of both the telomeric and centromeric copies are designated historically as exon 1, 2a, 2b, and 3-8. It is thought that gene conversion events may involve the two genes, leading to varying copy numbers of each gene. The protein encoded by this gene localizes to both the cytoplasm and the nucleus. Within the nucleus, the protein localizes to subnuclear bodies called gems which are found near coiled bodies containing high concentrations of small ribonucleoproteins (snRNPs). This protein forms heteromeric complexes with proteins such as SIP1 and GEMIN4, and also interacts with several proteins known to be involved in the biogenesis of snRNPs, such as hnRNP U protein and the small nucleolar RNA binding protein. Two transcript variants encoding distinct isoforms have been described.
FUNCTION: The SMN complex plays an essential role in spliceosomal snRNP assembly in the cytoplasm and is required for pre-mRNA splicing in the nucleus. It may also play a role in the metabolism of snoRNPs.
SUBUNIT STRUCTURE: Component of an import snRNP complex composed of KPNB1, RNUT1, SMN1 and ZNF259. Part of the core SMN complex that contains SMN1, SIP1/GEMIN2, DDX20/GEMIN3, GEMIN4, GEMIN5, GEMIN6, GEMIN7, GEMIN8 and STRAP/UNRIP. Interacts with DDX20, FBL, NOLA1, RNUT1, SYNCRIP and with several spliceosomal snRNP core Sm proteins, including SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE and ILF3. Interacts with OSTF1.
SUBCELLULAR LOCATION: Cytoplasm. Nucleus › gem. Note: Localized in subnuclear structures next to coiled bodies, called Gemini of Cajal bodies (Gems).
TISSUE SPECIFICITY: Expressed in a wide variety of tissues. Expressed at high levels in brain, kidney and liver, moderate levels in skeletal and cardiac muscle, and low levels in fibroblasts and lymphocytes. Also seen at high levels in spinal cord. Present in osteoclasts and mononuclear cells (at protein level).
INVOLVEMENT IN DISEASE: Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 1 (SMA1) [MIM:253300]. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Autosomal recessive forms are classified according to the age of onset, the maximum muscular activity achieved, and survivorship. The severity of the disease is mainly determined by the copy number of SMN2, a copy gene which predominantly produces exon 7-skipped transcripts and only low amount of full-length transcripts that encode for a protein identical to SMN1. Only about 4% of SMA patients bear one SMN1 copy with an intragenic mutation. SMA1 is a severe form, with onset before 6 months of age. SMA1 patients never achieve the ability to sit.
Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 2 (SMA2) [MIM:253550]. SMA2 is an autosomal recessive spinal muscular atrophy of intermediate severity, with onset between 6 and 18 months. Patients do not reach the motor milestone of standing, and survive into adulthood.
Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 3 (SMA3) [MIM:253400]. SMA3 is an autosomal recessive spinal muscular atrophy with onset after 18 months. SMA3 patients develop ability to stand and walk and survive into adulthood.
Defects in SMN1 are the cause of spinal muscular atrophy autosomal recessive type 4 (SMA4) [MIM:271150]. SMA4 is an autosomal recessive spinal muscular atrophy characterized by symmetric proximal muscle weakness with onset in adulthood and slow disease progression. SMA4 patients can stand and walk.
MISCELLANEOUS: The SMN gene is present in two highly homologous and functional copies (TelSMN/SMN1 and CenSMN/SMN2). The telomeric copy of SMN gene (TelSMN/SMN1) seems to be the SMA-determining gene while the centromeric copy seems unaffected.
SEQUENCE SIMILARILITIES: Belongs to the SMN family.
Contains 1 Tudor domain.
Molecular Weight
35 kDa was observed; however, the calculated molecular weight is 31.849 kDa.
Physicochemical Information
Dimensions
Materials Information
Toxicological Information
Safety Information according to GHS
Safety Information
Product Usage Statements
Quality Assurance
Evaluated by Western Blot in HeLa cell lysate.
Western Blot Analysis: 0.5 µg/ml of this antibody detected SMN on 10 µg of HeLa cell lysate.
Usage Statement
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.